
Enhanced photocatalytic degradation of methylene blue and adsorption of arsenic(iii) by reduced graphene oxide (rGO)–metal oxide (TiO2/Fe3O4) based nanocomposites†
Abstract
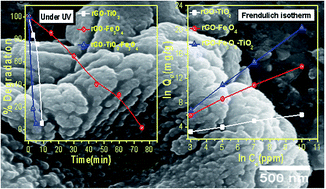
Reduced graphene oxide (rGO) and metal oxide based binary (rGO–TiO2/rGO–Fe3O4) and ternary (rGO–Fe3O4–TiO2) nanocomposites with enhanced photocatalytic and adsorption properties are successfully synthesized by a simple one-step solvothermal process. The microscopy images of the nanocomposites show that the ferric oxide (Fe3O4) and titania (TiO2) nanoparticles are firmly anchored over rGO, which enhances the surface area of the resultant nanocomposites. The as-synthesized nanocomposites are evaluated for the removal of methylene blue dye under UV and visible light irradiation as well as for the adsorption of As(III) from aqueous solution. Compared to binary, the ternary (rGO–Fe3O4–TiO2) nanocomposite exhibits the highest dye degradation efficiency (∼100% within 5 minutes). This enhancement is attributed to the synergetic interaction and increase in the surface area of rGO–Fe3O4–TiO2. For As(III) adsorption, the adsorption data are obtained by Langmuir and Freundlich adsorption isotherms. Compared to binary nanocomposites, the maximum monolayer adsorption capacity (147.05 mg g−1) is observed for rGO–Fe3O4–TiO2. These results reveal that the rGO–Fe3O4–TiO2 nanocomposite has potential application in water/wastewater treatment.
 Please wait while we load your content...
Please wait while we load your content...

