
The effect of amino substituents on the interactions of quinazolone derivatives with c-KIT G-quadruplex: insight from molecular dynamics simulation study for rational design of ligands†
Kiana Gholamjani Moghaddam and
Seyed Majid Hashemianzadeh*
Molecular Simulation Research Laboratory, Department of Chemistry, Iran University of Science & Technology, Tehran, Iran. E-mail: hashemianzadeh@iust.ac.ir; hashemianzadeh@gmail.com; Fax: +98 2177491204; Tel: +98 2177240287
First published on 4th September 2015
Abstract
Stabilization of G-quadruplex structures in the oncogenic promoter regions with small molecules has attracted considerable attention as a promising target for cancer therapy. To discover such small molecules, understanding the nature of interactions between the ligand and G-quadruplex is of paramount importance. To precisely investigate how these interactions can be influenced by varying different substituents, binding interactions of some quinazolone derivatives (QDs) with c-KIT G-quadruplex were studied by molecular dynamics (MD) simulation. The results revealed that the QD–NH–CO– arrangement in quinazolone derivatives improve binding affinity toward c-KIT G-quadruplex and the amino substituents play a crucial role in hydrogen bond formation and electrostatic interactions with the phosphate backbone of the G-quadruplex. We also proposed a new derivative of quinazolone (7k) with a terminal amino substituent instead of a 3-phenyl group. The binding free energy analysis suggested that this derivative stabilizes the c-KIT G-quadruplex much better than other derivatives. Furthermore, the calculated changes in solvent-accessible surface area (ΔSASA) were consistent with the binding free energy calculations. Our studies provide insight into the effect of different substituents on binding interactions between the ligand and G-quadruplex which can pave the way to rational ligand design.
1. Introduction
Guanine-rich sequences of DNA can be self-assembled into four-stranded structures defined as G-quadruplexes1–3 which are composed of stacked planar G-quartets, each of which is held together by a network of Hoogsteen hydrogen bonds between four guanine bases.4,5 G-quadruplex structures are mainly found in telomeres6 and particularly widespread in gene promoters such as k-ras,7 bcl2,8 c-myc,9 c-KIT,10,11 etc. It has been hypothesized that stabilization of G-quadruplex structures formed within the oncogene promoter regions with the aid of small molecules can attenuate gene overexpression and can be considered as a cancer therapeutic target.12–15The human c-KIT is a significant proto-oncogene that its expression is necessary to control different normal cells growth, whereas overexpression or mutation of this proto-oncogene has been noticed in various cancers including gastrointestinal stromal tumors (GIST), leukemias, melanoma and pancreatic cancers.16–20 Therefore, there is substantial interest in the design of small molecules that selectively stabilize c-KIT G-quadruplex which can result in inhibition of gene expression in cancer cells.
In recent years, many different ligands such as bisquinoline–pyrrole oligoamide,21 porphyrine,22 substituted indenoisoquinolines,23 benzo[a]phenoxazines,24 trisubstituted isoalloxazines,20 bis-indole carboxamides,25 biaryl polyamides,26 phenanthroline-2,9-bistriazoles,27 etc., have been synthesized and employed to stabilize G-quadruplex of the c-KIT promoter. Most of G-quadruplex stabilizing ligands follow structural features that contain a planar aromatic surface which stack on a G-quartet, cationic side chains which interact with the phosphate backbone, grooves and loops and also a cationic core in the center of the ligand which interact with the negatively charged center of G-quadruplex.28 In order to discover drug-like ligands which selectively bind to c-KIT G-quadruplex, Jia-Heng Tan's group synthesized a new class of quinazolone derivatives because these derivatives display broad-spectrum properties such as antibacterial, antidiabetic, anti-inflammatory, antitumor, and several others. These unfused aromatic ligands contain two cationic amino substituents and an expanded aromatic structure. The experimental studies showed that the variation of the substituents in quinazolone derivatives play a significant role on stabilizing c-KIT G-quadruplex.29 To understand how the substituents can influence the binding affinity, it is essential to investigate the nature and contribution of the interactions between these ligands and c-KIT G-quadruplex that has not yet been reported.
In this work, molecular docking and molecular dynamics (MD) simulation were performed to precisely study the nature of interactions between three quinazolone derivatives (7e, 7h and 7i with the same numbering as the experimental study) (Fig. 1) and c-KIT G-quadruplex. These three ligands, selected based on their structural features, are used to study the impact of their structural variations on the stability of c-KIT G-quadruplex. In ligand 7i, the role played by inflexible pyrrolidino group at the side chain of this ligand on the interactions between the ligand and c-KIT G-quadruplex was studied. The pyrrolidino group of ligand 7i was substituted by diethylamino group in ligand 7h to investigate the effect of flexibility of the side chain on G-quadruplex stabilization. Finally, amido bond (QD–CO–NH–) arrangement in ligand 7h was interconverted in ligand 7e to explore how the arrangement of QD–NH–CO can influence on binding affinity. The MD simulations provide a valuable insight into how the minor change from QD–CO–NH– in ligand 7h and 7i to QD–NH–CO– arrangement in ligand 7e can play a role in increasing electrostatic interactions and stabilizing c-KIT G-quadruplex.
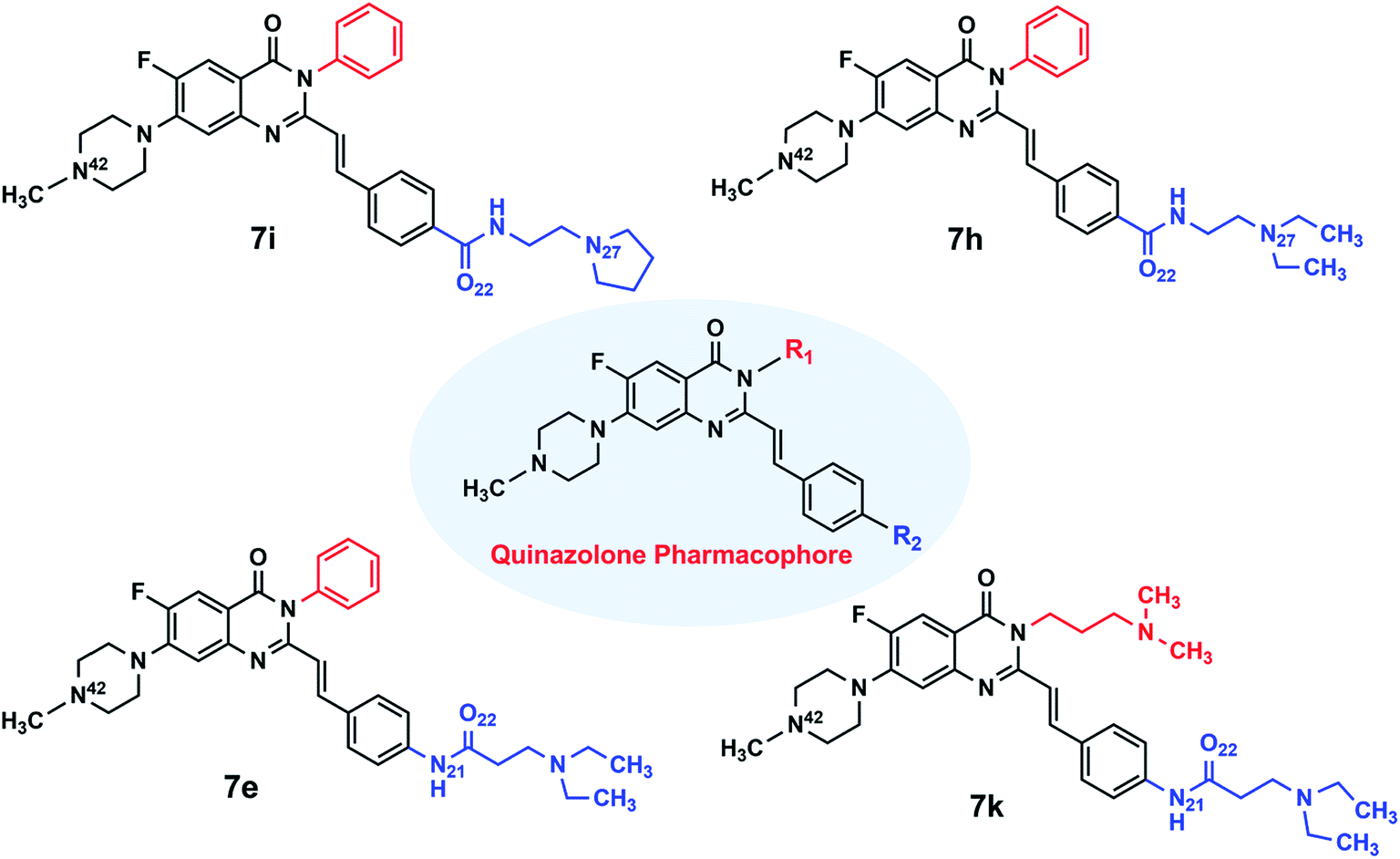 | ||
| Fig. 1 Quinazolone derivatives. | ||
Furthermore, molecular electrostatic potential maps (MEP) were plotted to support information about electrostatic interactions. In addition, Molecular Mechanics, Poisson–Boltzmann, Surface Area (MMPBSA) analysis was used to calculate binding free energies between these ligands and c-KIT G-quadruplex. The obtained results of free energy analysis provided valuable information regarding driving forces for G-quadruplex–ligand interaction and complex stability which are helpful in the design of potential ligands with high binding affinity. On this basis, we proposed a new derivative of quinazolone (7k) with a terminal amino side chain instead of 3-phenyl group. The aforementioned studies were carried out to explain in detail the effect of substituent modification on the binding of this ligand to c-KIT G-quadruplex.
2. Computational methods
For the molecular modeling studies, c-KIT-1 G-quadruplex (5′-AGGGAGGGCGCTGGGAGGAGGG-3′, PDB ID: 2O3M) was selected as a starting structure from the Protein Data Bank (PDB). The molecular structures of all ligands were optimized with DFT method at the hybrid functional B3LYP and the basis set 6-31+G(d) level using Gaussian 03 program.30 The partial charges were assigned using AM1-BCC method via the ACPYPE tool.31 Other parameters were generated based on the Generalized Amber Force Field (GAFF)32 using ANTECHAMBER module of AmberTools14.332.1. Molecular docking
The binding modes of the ligands with G-quadruplex were investigated with AUTODOCK 4.0 program.34 Nonpolar hydrogens were merged to their corresponding carbons. Gasteiger and Kollman partial charges were assigned for the ligands and G-quadruplex respectively by AutoDockTools (ADT). The generated ADT format files for the ligands and target G-quadruplex were utilized as an input for the AUTOGRID program. The dimensions of the grid box was set to 70 Å × 70 Å × 70 Å with 0.375 Å spacing at the center of the G-quadruplex structure. One hundred docking runs were performed for each complex. Lamarckian genetic algorithm (LGA) was used for each docking calculation. Maximum number of energy evaluations and population size were set to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 and 150 respectively. Other parameters were set as default. The results of docking calculations were analyzed using RMSD clustering based on 0.5 Å root mean square criterion.
500000 and 150 respectively. Other parameters were set as default. The results of docking calculations were analyzed using RMSD clustering based on 0.5 Å root mean square criterion.
2.2. Molecular dynamics (MD) simulation
MD simulations were carried out for the best conformers obtained from molecular docking studies using Gromacs 4.6.5 package35 with Parmbsc0 force field.36 For the stability of the G-quadruplex structure, two central K+ ions were located manually in the center of G-quartets. All complexes were solvated in a triclinic box of TIP3P37 water molecules with a 10.0 Å buffer between the molecule(s) periphery and box edge. Periodic boundary conditions were applied in all dimensions. 17K+ counter ions were added to G-quadruplex–ligand complexes to neutralize the systems. The particle mesh Ewald (PME) method was applied to calculate long-range electrostatic interactions38 and the LINCS algorithm39 was used to constrain all bonds. A cut-off of 10.0 Å was used for non-bonded van der Waals interactions. At first, the solvated structures were minimized about 2000 steps using the steepest descent algorithm. The systems were then equilibrated under NVT ensemble at 300 K during 200 ps. This was followed by a 300 ps NPT equilibration run at 1 bar pressure and 300 K temperature. The velocity rescaling algorithm was used for the temperature coupling at 300 K with a time constant 0.1 ps.40 The pressure was controlled via the Parrinello–Rahman barostat algorithm at 1 bar with time constant 1 ps.41,42 Finally, MD production run was performed for 50 ns under equivalent conditions at 1 bar and 300 K. The time step of 2.0 fs was applied in all MD simulations and the output files were saved every 2 ps for the further analysis. Finally, the results were analyzed and visualized by means of the Discovery Studio Visualizer 4.1 (http://accelrys.com/products/discovery-studio) and Chimera 1.10 (ref.43) programs.2.3. Molecular electrostatic potential (MEP)
Molecular electrostatic potentials were calculated at the B3LYP/6-31G+(d) optimized geometry for all ligands. For the G-quadruplex structures, MEP maps were generated using the Adaptive Poisson–Boltzmann Solver (APBS) program44 in AutoDockTools. The dielectric constant values for G-quadruplex and the surrounding solvent were set to 2 and 80 respectively. The dielectric boundary was defined around the molecular surface by setting the solvent radius to 1.4 Å.2.4. Free energy calculation
The binding free energies were calculated by MMPBSA (Molecular Mechanics, Poisson–Boltzmann, Surface Area) analysis45 using GMXAPBS tool.46,47 Note that explicit water molecules and counterions were removed from the MD trajectories whereas two bound K+ in the channel of G-quadruplex were considered in MMPBSA analysis as reported by Jayaram48 and Štefl et al.49For each snapshot, the MMPBSA calculated the binding free energy using the following equation:
| ΔGbind = Gcomplex − (Gligand + Gquadruplex) | (1) |
The free energy terms of the complex, ligand and G-quadruplex in above equation were calculated according to eqn (2):
| 〈G〉 = 〈EMM〉 + 〈Gsolv〉 − T〈SMM〉 | (2) |
T〈SMM〉 refers to the entropy term where T and S denote the temperature and entropy, respectively.
The molecular mechanics energy was obtained as the following sum:
| EMM = Eint + Ecoul + Evdw | (3) |
The solvation energy term in eqn (2) was estimated with the eqn (4):
| Gsolv = Gps + Gnps | (4) |
Gps and Gnps were calculated with Adaptive Poisson–Boltzmann Solver (APBS) program.
For the calculation of polar solvation term, dielectric constants of 1.0 and 80.0 were considered for the solute and solvent, respectively.
Nonpolar solvation term was calculated using the following equation:
| Gnps = γSASA + β | (5) |
2.5. Solvent accessible surface area (SASA) calculation
The solvent accessible surface area (SASA) was computed for each complex using g_sas tool of Gromacs. The accessible surface is generated by tracing the center of the probe sphere rolling along the van der Waals surface. A probe radius of 1.4 Å was used for the solvent. Total SASA for each molecule is sum of the hydrophobic and hydrophilic SASA. The calculations were performed by eqn (6):| ΔSASA = SASAcomplex − (SASAquadruplex + SASAligand) | (6) |
3. Result and discussion
3.1. Molecular docking
Molecular docking is a computer-based technique that is used to predict the possible binding sites between small molecule drug candidates and a receptor.50 In this study, molecular docking studies were carried out to predict the favorable binding mode of quinazolone derivatives with G-quadruplex. The results of clustering analysis revealed that all ligands stack at the 3′ end of G-quadruplex (Fig. S1 ESI†) and two protonated amino groups at the side chains of ligands interact with negatively charged backbone through hydrogen bonding and electrostatic interactions. The results of binding energies are listed in Table 1. The binding energies rank order is 7e > 7h > 7i that is consistent with experimental results.29 The result also emphasized that 7k shows slightly higher binding energy than other derivatives.| Complex | Binding energy (kcal mol−1) |
|---|---|
| 7e | −12.62 |
| 7h | −11.51 |
| 7i | −10.74 |
| 7k | −12.75 |
Finally, the lowest energy conformation of each complex was selected for molecular dynamics studies.
3.2. MD simulation
On the basis of docking results, 50 ns MD simulations were performed on all complexes. The final structure of each complex is shown in Fig. S2 (ESI†). To understand the nature of the interactions between the ligands and G-quadruplex, the MD simulation results were analyzed as follows.| Complex | Heavy atoms of G-quartet | Heavy atoms of backbone |
|---|---|---|
| 7e | 1.15 | 1.45 |
| 7h | 1.16 | 1.49 |
| 7i | 1.16 | 1.61 |
| 7k | 1.13 | 1.45 |
 | ||
| Fig. 2 Time dependence of the RMSDs (Å) of the heavy atoms in G-quartet (black) and backbone (red). | ||
As evident from Table 2, the RMSD values of G-quartet heavy atoms range from 1.13 to 1.16 Å, while the backbone of G-quadruplex has slightly higher RMSD fluctuations. This behavior is because of high rigidity of G-quartets which are held strongly by eight hydrogen bonds whereas the backbone residues are free to move over the course of the simulation.
 | ||
| Fig. 3 Hoogsteen hydrogen bonds in one G-quartet. | ||
| G-quartet | 7e | 7h | 7i | 7k |
|---|---|---|---|---|
| G-quartet 1 (G10–G13–G2–G6) | 99.5 | 99.5 | 99.5 | 99.6 |
| G-quartet 2 (G21–G14–G3–G7) | 97.0 | 96.4 | 96.4 | 97.0 |
| G-quartet 3 (G22–G15–G4–G8) | 99.5 | 99.3 | 99.1 | 99.7 |
According to Table 3, the average hydrogen bond occupancy of G-quartets is in the order of 7k > 7e > 7h > 7i. This means that the average hydrogen bond occupancy of G-quartets is slightly higher for 7e complex (98.7%) compared to 7h (98.4%) and 7i (98.3%) complexes, indicating that ligand 7e with the QD–NH–CO– arrangement has a stabilizing effect on G-quartets which is consistent with results reported from the experimental studies.29 Moreover, the average hydrogen bond occupancy of ligand 7k (98.8%) implies that this new derivative of quinazolone provides slightly more stability to G-quartets.
In addition, hydrogen bonding data between G-quadruplex and ligands were analyzed and summarized in Table 4. Note that in hydrogen bonding calculations, the cutoff distance and angle deviation were set to 3.5 Å and 35° respectively. As shown in detail in Table 3, ligand 7e and 7k (Fig. 4) with the QD–NH–CO– arrangement form three hydrogen bonds with G-quadruplex structure contributing to its stabilization. In this case, one hydrogen bond was formed between O4′ atom of G20 and protonated terminal tertiary amine (N42) with a mean distance of 3.5 Å and 3.0 Å for 7e and 7k, respectively. On the other hand, ligand 7e and 7k were hydrogen bonded via hydrogen atom (H22) connected with the nitrogen atom no. 21 (N21) to O1 atom of G17 phosphate backbone and O3′ atom of A16.
| Complex | Donor | Acceptor | Occupancy (%) | Distance (Å) | Angle (°) |
|---|---|---|---|---|---|
| 7e | 25LIG(H45)N42 | 20 DG (O4′) | 66.6 | 3.5(0.9) | 26(14) |
| 25LIG(H22)N21 | 17 DG (O1P) | 54.9 | 3.3(0.7) | 35(27) | |
| 25LIG(H22)N21 | 16 DA (O3′) | 63.9 | 3.2(0.6) | 32(26) | |
| 7h | 25LIG(H45)N42 | 20 DG (O4′) | 72.3 | 3.2(0.4) | 26(12) |
| 25LIG(H28)N27 | 17 DG (O1P) | 76.3 | 3.2(0.7) | 21(20) | |
| 7i | 25LIG(H45)N42 | 20 DG (O4′) | 91.3 | 3.0(0.2) | 22(8) |
| 25LIG(H28)N27 | 17 DG (O1P) | 87.6 | 3.0(0.5) | 19(21) | |
| 7k | 25LIG(H45)N42 | 20 DG (O4′) | 93.3 | 3.0(0.2) | 21(8) |
| 25LIG(H22)N21 | 17 DG (O1P) | 48.5 | 3.3(0.6) | 41(32) | |
| 25LIG(H22)N21 | 16 DA (O3′) | 66.9 | 3.1(0.4) | 32(27) |
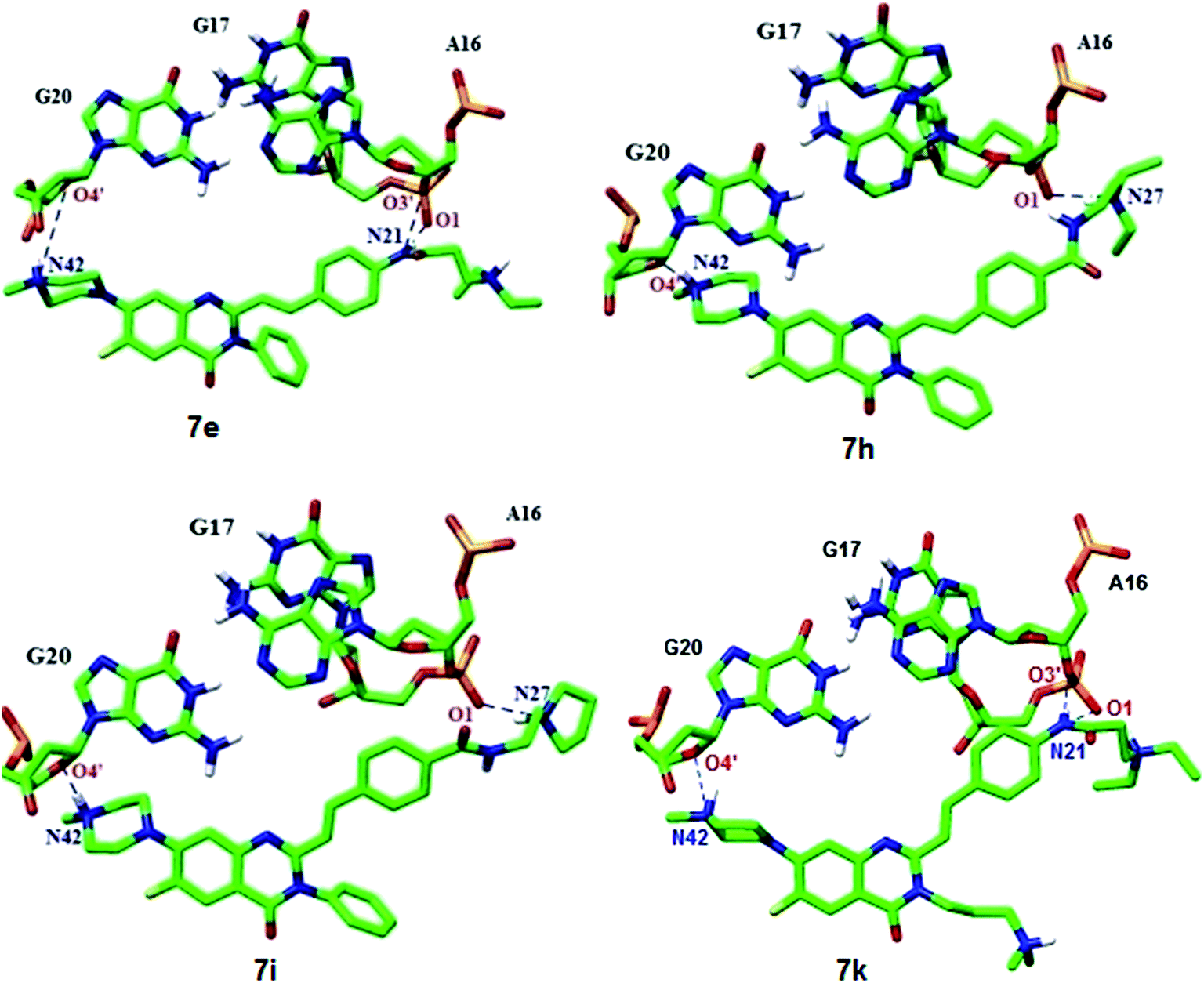 | ||
| Fig. 4 Hydrogen bonds formed from MD simulation for 7e, 7h, 7i and 7k complexes. | ||
As shown in Fig. 4, for ligand 7h and 7i with the QD–CO–NH– arrangement, two protonated terminal amines at the side chains (N42 and N27) were close to the O4′ atom of G20 and O1 atom of G17 to form the hydrogen bonding interactions. The mean hydrogen bond distances between these two amines and the backbone of G-quadruplex (O4′ atom of G20 and O1 atom of G17) are 3.2 Å (7h) and 3.0 Å (7i).
Therefore, it can be concluded that the formed hydrogen bonds between G-quadruplex and the ligands play an active role to enhance binding affinity of the ligands toward G-quadruplex.
 | ||
| Fig. 5 Electrostatic potential map of ligands. | ||
As can be seen from Fig. 5, the positive electrostatic potential areas for ligand 7k are mainly focused on the three terminal tertiary amines. Therefore, these amino groups are considered as active sites for the electrostatic interactions. It is worth noting that the introduction of a third amino group instead of 3-phenyl group provides more opportunities for electrostatic interactions with c-KIT G-quadruplex compared to other ligands, and as such increases complex stabilization.
Furthermore, MEPs for all complexes were mapped on the molecular surface after MD simulations. As shown in Fig. 6, the negative charge of G-quadruplex provides an overall negative electrostatic potential (colored red) which can increase its electrostatic interactions with the positively charged regions of the ligands.
 | ||
| Fig. 6 Electrostatic potential maps of G-quadruplex–ligand complexes. | ||
| Complex | ΔEcoul | ΔEvdw | ΔGps | ΔGnps | ΔGpolar | ΔGnonpolar | ΔGbind |
|---|---|---|---|---|---|---|---|
| a ΔEcoul, and ΔEvdw are the electrostatic and van der Waals energies. ΔGps and ΔGnps are the polar and nonpolar contributions to the solvation energy. ΔGpolar is sum of the electrostatic energy (ΔEcoul) and polar solvation energy (ΔGps). ΔGnonpolar is sum of the van der Waals energy (ΔEvdw) and nonpolar solvation energy (ΔGnps). ΔGbind is the binding free energy (ΔEcoul + ΔEvdw + ΔGps + ΔGnps). | |||||||
| 7e | −4289.352 | −238.151 | 4474.126 | −20.932 | 184.774 | −259.083 | −74.309 |
| 7h | −4157.282 | −237.727 | 4343.679 | −19.669 | 186.397 | −257.396 | −70.999 |
| 7i | −3591.523 | −192.934 | 3739.484 | −16.014 | 147.961 | −208.948 | −60.987 |
| 7k | −5930.451 | −238.484 | 6094.573 | −19.610 | 164.122 | −258.094 | −93.973 |
In order to obtain favorable contribution of each energy term, electrostatic energy (ΔEcoul), van der Waals energy (ΔEvdw), polar solvation energy (ΔGps) and nonpolar solvation energy (ΔGnps) were investigated for each complex. As can be seen from Table 5, the contributions of nonpolar solvation energy (ΔGnps) are slightly favorable for the interactions between ligands and G-quadruplex. On the contrary, the values of polar solvation energies (ΔGps) are very high positive and enormously unfavorable for binding. On the other hand, the comparison of total gas phase energies (ΔEcoul + ΔEvdw) and solvation energies revealed that electrostatic, van der Waals and nonpolar solvation energies are the predominant factors for the binding of ligands to G-quadruplex. The very high negative values of electrostatic energies (ΔEcoul) for all complexes implied that the protonated terminal amino groups at the side chains of ligands interact strongly with the anionic charges on the G-quadruplex backbone. In addition, the van der Waals energy contribution (ΔEvdw) is favorable for all ligands because the quinazolone and benzylidene pharmacophores make van der Waals contacts with G-quartet 3 surface as shown in Fig. S3 (ESI†).
The energy calculations exhibited the negative binding free energies for all complexes, indicating the G-quadruplex stability and binding affinity of quinazolone derivatives. As it can be realized from Table 5 and 7e complex is significantly more stable than 7h and 7i complexes which are in agreement with the reported experimental findings.29 Detailed energy analysis illustrated that ligand 7i with pyrrolidino group at the side chain shows higher binding free energy (ΔGbind = −60.987 kcal mol−1) compared to other ligands. This behavior is due to the rigidity of the pyrrolidino group and inaccessibility of the loop. To increase the flexibility of the side chain, the pyrrolidino group was substituted by diethylamino group in ligand 7h. The binding free energy calculations showed that this substitution enhances binding affinity and G-quadruplex stabilization (ΔGbind = −70.999 kcal mol−1). In order to investigate the effect of amido bond (QD–CO–NH–) arrangement on c-KIT G-quadruplex stabilization, we studied ligand 7e with interconverted amido bond. The results indicated that ligand 7e with QD–NH–CO– arrangement provides strong affinity with c-KIT G-quadruplex (ΔGbind = −74.309 kcal mol−1) when compared to those of 7h and 7i. The current findings suggest that the minor change from QD–CO–NH– (7h and 7i) to QD–NH–CO– (7e) arrangement plays a significant role in increasing electrostatic interactions and stabilizing c-KIT G-quadruplex.
The results of free energy calculations allowed us identifying electrostatic interactions as the main driving force for the binding between quinazolone derivatives and c-KIT G-quadruplex. In addition, MD simulation studies indicated that the phenyl group at the position 3 is not in plane with respect to the quinazolone and benzylidene pharmacophores of these ligands and as a result this group cannot stack on G-quartet surface. Therefore, the phenyl group did not play an active role in interaction with c-KIT G-quadruplex and binding affinity. Moreover, it has been reported that the amino side chains are positively charged at physiological pH which contribute to the strong electrostatic interaction with the phosphate backbone of G-quadruplex.51 This evidence prompted us to design a new quinazolone derivative (7k) which contain a terminal amino side chain instead of 3-phenyl group. We hypothesized that the amino group can enhance electrostatic interactions and G-quadruplex stabilization.
To investigate this hypothesize, the binding free energy of 7k complex was assessed in order to ascertain whether the ligand 7k has a stronger binding affinity to G-quadruplex compared with the other ligands. Surprisingly, the binding free energy results emphasized that 7k complex is considerably more stable than the other complexes due to its lowest binding free energy of −93.973 kcal mol−1. In fact, introduction of a terminal amino substituent instead of 3-phenyl group in ligand 7k dramatically improved electrostatic interactions which subsequently enhanced its binding affinity to c-KIT G-quadruplex.
| Complex | SASAcomplex | SASAG-quadruplex | SASAligand | ΔSASA |
|---|---|---|---|---|
| 7e | 4839.70 | 4530.49 | 824.28 | −515.07 |
| 7h | 4851.14 | 4524.53 | 818.60 | −491.99 |
| 7i | 4864.03 | 4506.13 | 813.23 | −455.33 |
| 7k | 4881.76 | 4528.89 | 872.45 | −519.58 |
4. Conclusions
Interactions of c-KIT G-quadruplex with four derivatives of quinazolone were studied by MD simulations to comparatively investigate the impact of substituent variations on the nature of binding. The energy calculations revealed that the van der Waals and electrostatic energies between c-KIT G-quadruplex and ligand 7h with diethylamino group was more favorable than ligand 7i with pyrrolidino group. As a result, the affinity of ligand 7h to G-quadruplex was higher than 7i. Nevertheless, the QD–CO–NH– arrangement in ligand 7h played a key role on stabilization ability of G-quadruplex. Our results demonstrated that the electrostatic interactions between c-KIT G-quadruplex and ligand 7h was enhanced by changing substituent arrangement from QD–CO–NH (7h) to QD–NH–CO (7e). These findings suggested that the contributions of the electrostatic interactions between c-KIT G-quadruplex and quinazolone derivatives are significantly more than other interactions. Based on the results, we introduced a new derivative of quinazolone (7k) with a terminal amino substituent instead of 3-phenyl group to increase the electrostatic interactions. The binding free energy analysis confirmed that the newly designed ligand (7k) interacts more strongly with c-KIT G-quadruplex compared to the other derivatives. Our computational studies help explain how the nature and contribution of interactions in G-quadruplex–ligand binding play crucial roles on G-quadruplex stabilization that directly influence designing of new potential ligands with high binding affinity.Acknowledgements
We would like to thank Dr. Sabzyan for providing us with hardware and software facilities.References
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402 CrossRef CAS PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2005, 33, 2908 CrossRef CAS PubMed.
- A. K. Todd, M. Johnston and S. Neidle, Nucleic Acids Res., 2005, 33, 2901 CrossRef CAS PubMed.
- S. Neidle, Curr. Opin. Struct. Biol., 2009, 19, 239 CrossRef CAS.
- S. Neidle and S. Balasubramanian, Quadruplex nucleic acids, RSC, Cambridge, U.K., 2006 Search PubMed.
- H. J. Lipps and D. Rhodes, Trends Cell Biol., 2009, 19, 414 CrossRef CAS PubMed.
- S. Cogoi and L. E. Xodo, Nucleic Acids Res., 2006, 34, 2536 CrossRef CAS PubMed.
- J. Dai, T. S. Dexheimer, D. Chen, M. Carver, A. Ambrus, R. A. Jones and D. Yang, J. Am. Chem. Soc., 2006, 128, 1096 CrossRef CAS PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593 CrossRef CAS PubMed.
- H. Fernando, A. P. Reszka, J. Huppert, S. Ladame, S. Rankin, A. R. Venkitaraman, S. Neidle and S. Balasubramanian, Biochemistry, 2006, 45, 7854 CrossRef CAS PubMed.
- S. Rankin, A. P. Reszka, J. Huppert, M. Zloh, G. N. Parkinson, A. K. Todd, S. Ladame, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2005, 127, 10584 CrossRef CAS PubMed.
- Z. A. Waller, S. A. Sewitz, S. T. D. Hsu and S. Balasubramanian, J. Am. Chem. Soc., 2009, 131, 12628 CrossRef CAS PubMed.
- J. Dash, P. S. Shirude, S. T. D. Hsu and S. Balasubramanian, J. Am. Chem. Soc., 2008, 130, 15950 CrossRef CAS PubMed.
- K. Jantos, R. Rodriguez, S. Ladame, P. S. Shirude and S. Balasubramanian, J. Am. Chem. Soc., 2006, 128, 13662 CrossRef CAS PubMed.
- M. Bejugam, S. Sewitz, P. S. Shirude, R. Rodriguez, R. Shahid and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 12926 CrossRef CAS PubMed.
- S. Sakurai, T. Fukasawa, J. M. Chong, A. Tanaka and M. Fukayama, Cancer Sci., 1999, 90, 1321 CrossRef CAS PubMed.
- A. Yasuda, H. Sawai, H. Takahashi, N. Ochi, Y. Matsuo, H. Funahashi, M. Sato, Y. Okada, H. Takeyama and T. Manabe, Mol. Cancer, 2006, 5, 46 CrossRef PubMed.
- A. McIntyre, B. Summersgill, B. Grygalewicz, A. J. Gillis, J. Stoop, R. J. van Gurp, N. Dennis, C. Fisher, R. Huddart and C. Cooper, et al., Cancer Res., 2005, 65, 8085 CrossRef CAS PubMed.
- M. Miettinen and J. Lasota, Appl. Immunohistochem. Mol. Morphol., 2005, 13, 205 CrossRef CAS.
- K. S. Smalley, K. L. Nathanson and K. T. Flaherty, Cancer Res., 2009, 69, 3241 CrossRef CAS PubMed.
- C. W. Ong, M. C. Liu, K. D. Lee, K. W. Chang, Y. T. Yang, H. W. Tung and K. R. Fox, Tetrahedron, 2012, 68, 5453 CrossRef CAS PubMed.
- S. Manaye, R. Eritja, A. Aviñó, J. Jaumot and R. Gargallo, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 1987 CrossRef CAS PubMed.
- M. Bejugam, M. Gunaratnam, S. Müller, D. A. Sanders, S. Sewitz, J. A. Fletcher, S. Neidle and S. Balasubramanian, ACS Med. Chem. Lett., 2010, 1, 306 CrossRef CAS PubMed.
- K. I. McLuckie, Z. A. Waller, D. A. Sanders, D. Alves, R. Rodriguez, J. Dash, G. J. McKenzie, A. R. Venkitaraman and S. Balasubramanian, J. Am. Chem. Soc., 2011, 133, 2658 CrossRef CAS PubMed.
- J. Dash, R. Nath Das, N. Hegde, G. D. Pantoş, P. S. Shirude and S. Balasubramanian, Chem.–Eur. J., 2012, 18, 55 CrossRef PubMed.
- K. M. Rahman, A. P. Reszka, M. Gunaratnam, S. M. Haider, P. W. Howard, K. R. Fox, S. Neidle and D. E. Thurston, Chem. Commun., 2009, 4097 RSC.
- M. C. Nielsen, A. F. Larsen, F. H. Abdikadir and T. Ulven, Eur. J. Med. Chem., 2014, 72, 119 CrossRef CAS PubMed.
- J. E. Reed, A. A. Arnal, S. Neidle and R. Vilar, J. Am. Chem. Soc., 2006, 128, 5992 CrossRef CAS PubMed.
- X. Wang, C. X. Zhou, J. W. Yan, J. Q. Hou, S. B. Chen, T. M. Ou, L. Q. Gu, Z. S. Huang and J. H. Tan, ACS Med. Chem. Lett., 2013, 4, 909 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven Jr, K. N. Kudin and J. C. Burant, et al., Gaussian 03, Gaussian, Inc, Wallingford, CT, 2004 Search PubMed.
- A. W. Sousa da Silva and W. F. Vranken, BMC Res. Notes, 2012, 23, 367 CrossRef PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701 CrossRef CAS PubMed.
- A. Pérez, I. Marchán, D. Svozil, J. Sponer, T. E. Cheatham, C. A. Laughton and M. Orozco, Biophys. J., 2007, 92, 3817 CrossRef PubMed.
- D. J. Price and C. L. Brooks III, J. Chem. Phys., 2004, 121, 10096 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182 CrossRef CAS.
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055 CrossRef.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889 CrossRef CAS PubMed.
- D. Spiliotopoulos, A. Spitaleri and G. Musco, PLoS One, 2012, 7, e46902 CAS.
- C. Paissoni, D. Spiliotopoulos, G. Musco and A. Spitaleri, Comput. Phys. Commun., 2014, 186, 105 CrossRef PubMed.
- B. Jayaram, D. Sprous, M. A. Young and D. L. Beveridge, J. Am. Chem. Soc., 1998, 120, 10629 CrossRef CAS.
- R. Štefl, T. E. Cheatham, N. A. Špačková, E. Fadrná, I. Berger, J. Koča and J. Šponer, Biophys. J., 2003, 85, 1787 CrossRef.
- S. Alcaro, C. Musetti, S. Distinto, M. Casatti, G. Zagotto, A. Artese, L. Parrotta, F. Moraca, G. Costa, F. Ortuso, E. Maccioni and C. Sissi, J. Med. Chem., 2013, 56, 843 CrossRef CAS.
- T. M. Ou, Y. J. Lu, C. Zhang, Z. S. Huang, X. D. Wang, J. H. Tan, Y. Chen, D. L. Ma, K. Y. Wong, J. C. O. Tang, A. S. C. Chan and L. Q. Gu, J. Med. Chem., 2007, 50, 1465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra13615f |
| This journal is © The Royal Society of Chemistry 2015 |