
Metal oxide nanostructures incorporated/immobilized paper matrices and their applications: a review
Abstract
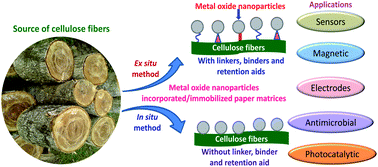
Metal oxide nanostructures of TiO2, ZnO, Fe2O3/Fe3O4, Bi2O3, CeO2, ITO, SiO2, MoO2, and WO3 have shown great potential for applications in various fields such as piezoelectric, magnetic, gas sensors, and dye sensitized solar cells due to their unique optical, electronic, conductivity, catalytic and antimicrobial properties. However, recovery and reuse of these nanostructures pose big threats from cost and environmental perspectives. Thus, various substrates have been employed for incorporating or immobilizing them but finding suitable substrate is still a big challenge. Paper, being a natural biopolymer, has been used recently to incorporate/immobilize various metal oxide nanostructures. The metal oxide nanostructures are normally adhered to the cellulose matrices through weak interactions such as van der Waals force and hence, usually have retention related issues. This was circumvented to a great extent by using suitable linkers, binders, or retention aids for the incorporation/immobilization of the nanostructures in paper matrices. Although these reagents improve retention, as well as some of the properties, they ultimately add cost to the final product. Additionally, these retention aids and linkers hinder accessibility of active surface sites of metal oxide nanostructures for their various applications. Very recently our group developed an in situ single step hydrothermal method to immobilize metal oxide nanostructures such as TiO2, ZnO, and Bi2O3 without using any binder, linker or retention aid. In this review a comprehensive account of the development of methodology for incorporation/immobilization of metal oxide nanostructures is discussed. Furthermore, how the immobilization of nanostructures evolved without using any binder, linker or retention aid is thoroughly discussed based on the chemistry of the cellulose and metal oxides. Applications of nanostructure immobilized paper matrices are highlighted and critical challenges are discussed along with directions for future research.
 Please wait while we load your content...
Please wait while we load your content...

