
Convenient synthesis of organic-electronics-oriented building blocks via on-water and under-air homocoupling of (hetero)aryl iodides†
Yi-An Chen and
Ching-Yuan Liu*
Department of Chemical and Materials Engineering, National Central University, Jhongli District, Taoyuan, Taiwan 320, Republic of China. E-mail: cyliu0312@ncu.edu.tw; Tel: +886-3-422-7151
First published on 25th August 2015
Abstract
We report herein an operationally simple homocoupling reaction that targets the convenient synthesis of organic-electronically important building blocks. A variety of synthetically useful bithiophene derivatives and functionalized biphenyls are efficiently prepared by an on-water and under-air protocol using Pd/C as catalyst. We find that Pd/C gives generally higher and cleaner homocoupling conversions than using Pd(OAc)2 in the cases of (hetero)aryl iodides since Pd(OAc)2 triggers more side reactions including dehalogenations and oligomerizations. Under the optimum conditions, a broad range of functional groups such as ester, ketone, aldehyde, nitrile, nitro, chloride, and bromide are well tolerated. We expect the present methodology would make a valuable synthetic contribution towards bridging green chemistry with thiophene-based organic materials.
Introduction
The transition-metal-catalyzed homocoupling reaction is one of the most convenient and reliable synthetic tools for the facile construction of C(sp2)–C(sp2) bonds.1 The resulting symmetrical bi(hetero)aryls are essential building blocks for numerous bioactive compounds2 and organic optoelectronic materials.3 Focusing on the field of organic electronics, bithiophene-containing molecules constitute an important class of π-functional semiconducting materials, for instance, bithiophene and its alkylated derivatives were often incorporated into oligoaryls as π-spacers or utilized as electron-donating part for donor–acceptor (D–A) type copolymers, both of which have been extensively applied in OPVCs,4 DSSCs,5 and OFETs.6 Although the bithiophene moiety is of substantial impact on modern materials science, synthetic methods reported to date to access it usually rely on the [Cu]- or [Pd]-catalyzed homocouplings of halogenated or metalated thiophenes in organic phases7,8 (eqn (1) and (2), Scheme 1). Despite some reports have disclosed the Pd-catalyzed homocoupling of aryl halides using water as reaction solvent,9 the synthetic focus has never placed on the preparation of organic-electronically versatile bithiophene derivatives. Therefore, we demonstrate herein a Pd-catalyzed on-water and under-air homocoupling methodology that aims to efficiently and environment-friendly synthesize bi(hetero)aryls including various alkylated bithiophenes that are extensively used as versatile building blocks for π-functional materials (Scheme 1). | ||
| Scheme 1 Synthetically versatile bithiophene derivatives prepared by homocoupling reactions conducted in organic phase or in aqueous phase. | ||
Results and discussion
To obtain the optimum reaction conditions, different palladium sources, reducing agents, and solvents were screened, as shown in Table 1, using the readily synthesized 3-hexyl-2-iodothiophene (1a), an important and representative monomer in polymer chemistry, as homocoupling substrate. In the preliminary experiments (entries 1 and 2), it was found that the addition of tetrabutylammonium bromide (TBAB) as surfactant was essential to the success of the on-water type reactions. In the absence of TBAB, the homocoupling did not take place (entry 1), whereas the desired product (2a) was isolated in 51% yield when 2 equiv. of TBAB was added (entry 2). More importantly, in addition to the homocoupled adduct, we have observed, under the catalysis of Pd(OAc)2, the formation of undesired deiodinated byproducts and some oligomeric hexylthiophenes that were generated through direct C–H heteroarylation10 of 1a, both of which are competitive with the formation of 2a and led to the diminishment of reaction conversion. Therefore, we turned to examine the Pd(0)-based catalysts. In contrast to the results obtained by using Pd(OAc)2, it is worth noting that formation of both byproducts (deiodinated and oligomerized thiophenes) was effectively suppressed with the employment of all Pd(0)-catalysts (entries 3–5). We were pleased to find that Pd/C gave a promising yield (80%, entry 5) and there was no significant yield variation when half amount of Pd/C was loaded (77%, entry 6). Next on, the solvent effect was investigated and the results revealed that using cosolvent systems did not lead to expected improvement of reaction conversion and 2a was isolated in poor to moderate yields (10–63%, entries 7–10). Finally, we tested different reducing agents including zinc, manganese, and copper, affording product 2a in 27–50% yields (entries 11–13). Without the addition of either reducing agent or catalyst, the homocoupling reaction did not occur and starting material was recovered (entries 14–15). As expected, the bromo-congener of 1a was shown to exhibit poor reactivity and compound 2a was isolated in only 33% yield (entry 16).
|
||||
|---|---|---|---|---|
| Entry | [Pd] (mol%) | Reducing agent | Solvent | Yieldf (%) |
a Unless otherwise noted, the homocoupling reaction was performed with 3-hexyl-2-iodothiophene 1a (1.0 mmol) in the presence of [Pd] (5–10 mol%), reducing agents (0.5 mmol), lithium chloride (1.5 mmol), and tetrabutylammonium bromide (TBAB, 2.0 mmol) using water as solvent (2.0 mL) at 90 °C for 24 h under air.b TBAB was not added.c A cosolvent system is used v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.d Indium powder and lithium chloride were not added.e The bromo-congener of compound 1a (2-bromo-3-hexylthiophene) was used.f Isolated yields. :1.d Indium powder and lithium chloride were not added.e The bromo-congener of compound 1a (2-bromo-3-hexylthiophene) was used.f Isolated yields. |
||||
| 1b | Pd(OAc)2 (10) | In | H2O | 0 |
| 2 | Pd(OAc)2 (10) | In | H2O | 51 |
| 3 | Pd2dba3 (10) | In | H2O | 56 |
| 4 | Pd(dba)2 (10) | In | H2O | 67 |
| 5 | Pd/C (10) | In | H2O | 80 |
| 6 | Pd/C (5) | In | H2O | 77 |
| 7c | Pd/C (5) | In | H2O/t-BuOH | 10 |
| 8c | Pd/C (5) | In | H2O/acetone | 63 |
| 9c | Pd/C (5) | In | H2O/DMA | 21 |
| 10c | Pd/C (5) | In | H2O/DMSO | 58 |
| 11 | Pd/C (5) | Zn | H2O | 28 |
| 12 | Pd/C (5) | Mn | H2O | 27 |
| 13 | Pd/C (5) | Cu | H2O | 50 |
| 14d | Pd/C (5) | ─ | H2O | 0 |
| 15 | ─ | In | H2O | 0 |
| 16e | Pd/C (5) | In | H2O | 33 |

Encouraged by the optimal reaction conditions obtained in Table 1, we then tried to explore the substrate scope of present homocoupling reactions using inexpensive and nontoxic water as sole solvent. As demonstrated in Table 2, we firstly focused on the preparation of synthetically and organic-electronically useful biheteroaryls including various (alkyl)bithiophenes and their related derivatives. A typical bithiophene molecule was obtained in 70% yield (2b). Further, a serious of bithiophene derivatives bearing different alkyl chains were efficiently synthesized in moderate to good yields (48–83%, 2c–2h). Interestingly, unprecedented examples such as bithiophenes possessing alkyl chains with a phenyl or an ester group were successfully prepared for the first time by this on-water methodology and isolated in moderate yields (52–66%, 2i–2j). Next on, since we had observed the poor reactivity of 2-bromo-3-hexylthiophene (entry 16, Table 1), we anticipated that a good chemoselectivity would be obtained when using 3-bromo-2-iodothiophene as homocoupling substrate. However, desired product 2k was isolated in only 32% yield and a significant amount of starting material (1k) was recovered, presumably owing to the steric hindrance caused by the 3-positioned bromine atom. In contrast, we acquired an improved yield when an unhindered regioisomer of 1k was employed (1l: 2-bromo-5-iodothiophene), giving the corresponding product 2l in 61% yield. Additionally, a number of alkyl-terminated bithiophenes were readily synthesized under identical reaction conditions, affording compounds 2m–2q in moderate to good yields (45–77%). Bis-ethylenedioxythiophene (bis-EDOT), an electron-rich derivative of thiophenes, was prepared with a relatively lower isolated yield (36%, 2r) because in this case the byproduct resulted from deiodination was formed and it was difficult to be separated from desired bis-EDOT. Finally, we were pleased to find that biselenophene could be also produced via present synthetic approach (50%, 2s).


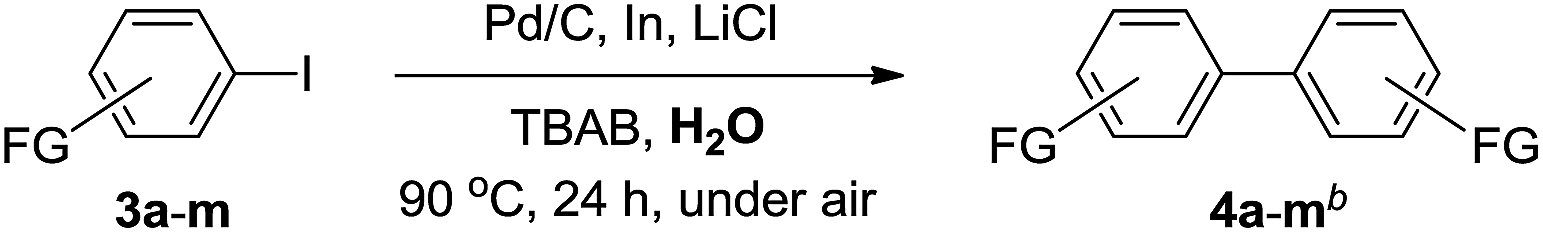
In order to further extend substrate scope and examine functional groups (FGs) compatibility of this on-water homocoupling reaction, aryl iodides bearing a broad range of synthetically useful FGs (3a–3m) were tested under optimal conditions (Table 3). The results have shown that sensitive FGs including ester, ketone, aldehyde, nitrile, nitro group, and halides were well tolerated and the corresponding biaryls were obtained in moderate to excellent isolated yields (45–92%, 4a–4m). It was found that the ortho-substituted iodoarenes (3c, 3e, and 3g) gave generally lower yields than their meta- or para-isomers (3a–b, 3d, and 3f), which is attributed to the anticipated steric hindrance caused by ortho substituents. More importantly, as discussed in Table 1, we also observed in each example a large quantity of deiodinated byproducts when the homocoupling reaction was catalyzed by Pd(OAc)2, whereas Pd/C was found to give more efficient and cleaner reactions with the generation of only trace amount of deiodinated compounds. Thus, it seems reasonable to conclude that, for current on-water homocoupling reactions, Pd/C would be a better alternative catalyst than the commonly used Pd(OAc)2. Moreover, Pd/C is especially suitable for the thiophene-based substrates with arylable C–H bonds because under Pd/C-catalysis the target reaction (homocoupling) proceeds smoothly while the undesired C–H oligomerization is inhibited.
As shown in Scheme 2, a plausible reaction mechanism for the Pd-catalyzed homocoupling is proposed using iodothiophenes (A) as substrate. Pd(0) undergoes oxidative addition with A to give a Pd(II) species (B). Reduction of B by indium/lithium halide affords an anionic thienylpalladium intermediate (C). Second oxidative addition of C with A proceeds to generate a dithienylpalladium(II) intermediate (D). Reductive elimination of D yields the desired bithiophene derivatives (E) and regenerates Pd(0).
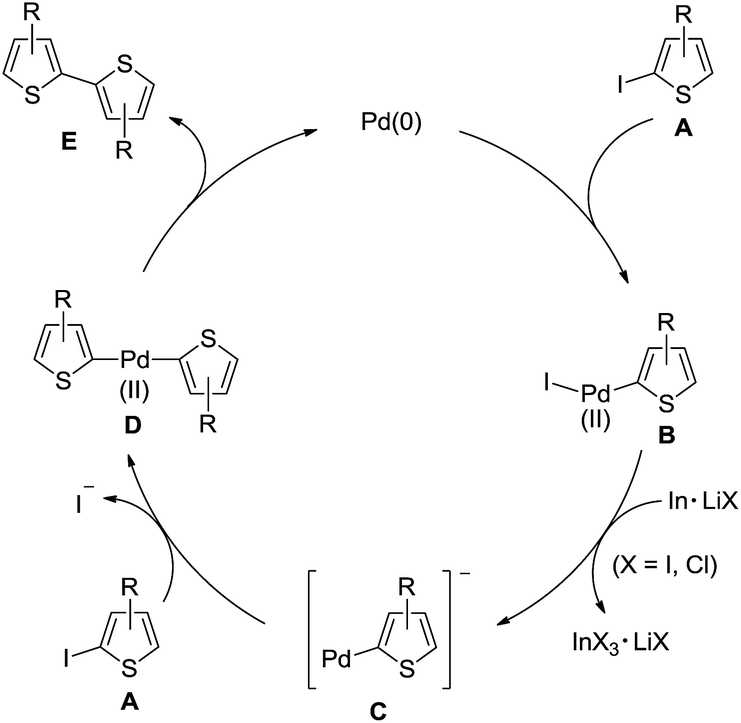 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed an on-water and under air homocoupling methodology targeting on the convenient synthesis of various organic-electronically important bithiophene derivatives. In this report, we found that, compared with Pd(OAc)2, using Pd/C as catalyst enabled a higher conversion of the homocoupled adduct because formation of the byproducts such as deiodinated or oligomeric thiophenes was inhibited. Under optimum reaction conditions, a variety of functional groups including ester, ketone, aldehyde, nitrile, nitro, and halides were well tolerated in the homocouplings of aryl iodides. We expect this convenient and viable on-water synthesis would become an user-friendly alternative route for polymer chemists to access various dialkyl-bithiophenes. Further synthetic investigation using water as solvent for functional π-conjugated organic materials is currently underway in our laboratory.Experimental section
General information
Unless otherwise specified, all reactions were carried out with magnetic stirring and, if air or moisture sensitive, in flame-dried glassware under nitrogen. Starting materials including the alkylated thiophene derivatives were synthesized according to the reported procedures.11 Reagents including a part of the (hetero)aryl iodides (1b, 3a–3m), palladium catalysts, ligands, and other additives are commercially available. In addition to the deionized water from Elix Millipore, organic solvents used in Table 1 such as acetone, tbutyl alcohol, tetrahydrofuran (THF), chloroform, and toluene were purchased from Sigma-Aldrich or Acros and used directly without further purifications. Syringes used to transfer reagents and solvents were purged with nitrogen prior to use. Reactions were monitored by thin layer chromatography (TLC, aluminum plates coated with silica gel, Merck 60, F-254). The spots were visualized by UV light. Flash column chromatography was performed using silica gel 60 (spherical, 63–210 μm) from Merck. The diameters of the columns and the amount of silica gel loaded were calculated according to the recommendation of W. C. Still.12 Melting points were measured on a Fargo MP-2D apparatus. NMR spectra were recorded on a Bruker Magnet System 300 or 500 MHz instrument. Chemical shifts were given relative to CDCl3 (7.26 ppm for 1H NMR, 77.0 ppm for 13C NMR), CD2Cl2 (5.32 ppm for 1H NMR, 54.0 ppm for 13C NMR), DMSO-d6 (2.50 ppm for 1H NMR, 39.4 ppm for 13C NMR), acetone-d6 (2.04 ppm for 1H NMR, 29.3 ppm for 13C NMR). For the characterization of the observed signal multiplicities, the following abbreviations were applied: s (singlet), d (doublet), dd (doublet of doublets), dt (doublet of triplets), dm (doublet of multiplets), t (triplet), td (triplet of doublets), q (quartet), quint (quintet), m (multiplet), comp (complex), app (apparent), and br (broad). Mass spectra were recorded on a JEOL JMS-700 for electron impact ionization (EI) and high resolution mass spectra (HRMS) on a JEOL JMS-700 spectrometers. Fast atom bombardment (FAB) samples were recorded in a 3-nitrobenzyl alcohol- or glycerine-matrix.General procedure A for Table 1: reaction conditions optimization of the on-water homocoupling reactions
To a heterogeneous solution of palladium catalysts (0.05–0.10 mmol), reducing agents (0.50 mmol), lithium chloride (1.50 mmol), and tetrabutylammonium bromide (TBAB) (2.00 mmol) in water (2 mL) or cosolvents (2 mL, v/v = 1:1) was added 3-hexyl-2-iodothiophene (1a) (1.00 mmol) under air. The reaction mixture was then heated at 90 °C for 24 h. After the reaction mixture had cooled to room temperature, the precipitate/solid was filtered off under vacuum and washed with ethyl acetate. The bi-phase filtrate was extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with brine (60 mL), and dried over Na2SO4 before concentrated in vacuo. Purification by flash chromatography yielded the desired product 2a.
General procedure B for Table 2: on-water homocoupling reactions of various heteroaryl iodides
To a heterogeneous solution of palladium on activated charcoal (Pd/C, 10% Pd basis, 0.05 mmol), indium powder (0.50 mmol), lithium chloride (1.50 mmol), and tetrabutylammonium bromide (TBAB) (2.00 mmol) in water (2 mL) was added the corresponding heteroaryl iodides (1b–1s) (1.00 mmol) under air. The reaction mixture was then heated at 90 °C for 24 h. After cooled to room temperature, the precipitate/solid was filtered off under vacuum and washed with ethyl acetate. The bi-phase filtrate was extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with brine (60 mL), and dried over Na2SO4 before concentrated in vacuo. Purification by flash chromatography yielded the desired products 2b–2s.:98) the pure product 2i (105 mg, 52%). A yellow liquid. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.05–7.42 (comp, 12H), 6.97 (d, J = 5.2 Hz, 2H), 2.56 (t, J = 7.5 Hz, 8H), 1.76–1.97 (comp, 4H); 13C NMR (CDCl3, 75 MHz, ppm): δ 142.2, 141.7, 128.9, 128.5, 128.3, 128.2, 125.7, 125.5, 35.6, 32.3, 28.5; MS (EI, 70 eV): 402 (M+, 100%), 298 (9%), 207 (16%), 193 (59%), 173 (28%), 91 (94%); HRMS (EI): calcd for C26H26S2: 402.1476, found: 402.1483.:80) the pure product 2j (148 mg, 66%). A yellow liquid. 1H NMR (CD2Cl2, 300 MHz, ppm): δ 7.29 (d, J = 5.3 Hz, 2H), 6.96 (d, J = 5.3 Hz, 2H), 4.05 (q, J = 7.1 Hz, 4H), 2.50 (t, J = 7.8 Hz, 4H), 2.23 (t, J = 7.5 Hz, 4H), 1.46–1.64 (comp, 8H), 1.10–1.32 (comp, 10H); 13C NMR (CDCl3, 75 MHz, ppm): δ 173.6, 141.9, 128.7, 128.4, 125.3, 60.1, 34.2, 30.3, 28.8, 28.5, 24.7, 14.2; MS (EI, 70 eV): 450 (M+, 96%), 193 (100%), 179 (46%), 147 (50%), 55 (52%); HRMS (EI): calcd for C24H34O4S2: 450.1899, found: 450.1893.:80) the pure product 2r (51 mg, 36%). A greenish yellow solid; m.p.: 204.1–205.9 °C. 1H NMR (CD2Cl2, 300 MHz, ppm): δ 6.26 (s, 2H), 4.18–4.36 (comp, 8H); 13C NMR (CD2Cl2, 75 MHz, ppm): δ 141.9, 137.7, 110.3, 97.9, 65.7, 65.3.General procedure C for Table 3: on-water homocoupling reactions of various functionalized aryl iodides
To a heterogeneous solution of palladium on activated charcoal (Pd/C, 10% Pd basis, 0.05 mmol), indium powder (0.50 mmol), lithium chloride (1.50 mmol), and tetrabutylammonium bromide (TBAB) (2.00 mmol) in water (2 mL) was added the corresponding aryl iodides (3a–3m) (1.00 mmol) under air. The reaction mixture was then heated at 90 °C for 24 h. After cooled to room temperature, the precipitate/solid was filtered off under vacuum and washed with ethyl acetate. The bi-phase filtrate was extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with brine (60 mL), and dried over Na2SO4 before concentrated in vacuo. Purification by flash chromatography yielded the desired product 4a–4m.:70) the pure product 4a (95 mg, 70%). A white solid; m.p.: 210.0–211.9 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.13 (d, J = 8.2 Hz, 4H), 7.69 (d, J = 8.2 Hz, 4H), 3.95 (s, 6H); 13C NMR (CDCl3, 75 MHz, ppm): δ 166.8, 144.3, 130.2, 129.7, 127.2, 52.2.:70) the pure product 4b (99 mg, 73%). A white solid; m.p.: 104.2–105.0 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.31 (s, 2H), 8.05 (d, J = 7.8 Hz, 2H), 7.82 (d, J = 7.8 Hz, 2H), 7.54 (t, J = 7.8 Hz, 2H), 3.96 (s, 6H); 13C NMR (CDCl3, 75 MHz, ppm): δ 166.8, 140.3, 131.4, 130.8, 128.9, 128.7, 128.2, 52.1.:70) the pure product 4c (70 mg, 52%). A white solid; m.p.: 71.9–72.4 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.01 (dd, J = 7.7, 1.4 Hz, 2H), 7.49–7.58 (comp, 2H), 7.37–7.47 (comp, 2H), 7.21 (dd, J = 7.7, 1.1 Hz, 2H), 3.62 (s, 6H); 13C NMR (CDCl3, 75 MHz, ppm): δ 167.4, 143.2, 131.4, 130.2, 129.8, 129.3, 127.1, 51.7.:70) the pure product 4d (82 mg, 69%). A white solid; m.p.: 192.4–193.2 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.05 (d, J = 8.2 Hz, 4H), 7.71 (d, J = 8.2 Hz, 4H), 2.64 (s, 6H); 13C NMR (CDCl3, 75 MHz, ppm): δ 197.5, 144.2, 136.5, 128.9, 127.4, 26.6.:70) the pure product 4e (54 mg, 45%). A white solid; m.p.: 91.2–93.0 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.73 (dd, J = 7.3, 1.5 Hz, 2H), 7.36–7.61 (comp, 4H), 7.17 (dd, J = 6.3, 1.3 Hz, 2H), 2.26 (s, 6H); 13C NMR (CDCl3, 75 MHz, ppm): δ 201.5, 140.6, 138.7, 131.0, 130.7, 128.5, 127.6, 29.2.:80) the pure product 4f (97 mg, 92%). A white solid; m.p.: 146.0–147.8 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 10.06 (s, 2H), 7.98 (d, J = 8.2 Hz, 4H), 7.78 (d, J = 8.2 Hz, 4H); 13C NMR (CDCl3, 75 MHz, ppm): δ 191.6, 145.4, 135.9, 130.3, 127.9.:80) the pure product 4g (79 mg, 75%). A yellow solid; m.p.: 58.5–60.0 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 9.84 (s, 2H), 8.06 (d, J = 7.3 Hz, 2H), 7.73–7.54 (comp, 4H), 7.35 (d, J = 7.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz, ppm): δ 191.0, 141.2, 134.6, 133.4, 131.7, 128.8, 128.5.:70) the pure product 4h (55 mg, 54%). A white solid; m.p.: 175.2–177.0 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.80–7.88 (comp, 2H), 7.68–7.76 (comp, 2H), 7.52–7.64 (comp, 4H); 13C NMR (CDCl3, 75 MHz, ppm): δ 141.5, 133.5, 132.8, 130.5, 129.2, 117.5, 112.3.:80) the pure product 4i (87 mg, 71%). A yellow solid; m.p.: 235.7–237.0 °C. 1H NMR (CDCl3, 300 MHz, ppm): δ 8.35 (d, J = 8.4 Hz, 4H), 7.79 (d, J = 8.4 Hz, 4H); 13C NMR (CDCl3, 75 MHz, ppm): δ 148.1, 145.0, 128.3, 124.4.Acknowledgements
Financial support provided by the Ministry of Science and Technology (MOST), Taiwan (MOST 103-2113-M-008-009-MY2) and the National Central University (NCU) are gratefully acknowledged.References
- (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed; (b) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed; (c) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed; (d) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (e) J. Hashim and C. O. Kappe, Adv. Synth. Catal., 2007, 349, 2353–2360 CrossRef CAS PubMed; (f) B. Kaboudin, Y. Abedi and T. Yokomatsu, Eur. J. Org. Chem., 2011, 6656–6662 CrossRef CAS PubMed; (g) D. H. Ortgies, F. Chen and P. Forgione, Eur. J. Org. Chem., 2014, 3917–3922 CrossRef CAS PubMed.
- (a) A. P. Degnan and A. I. Meyers, J. Am. Chem. Soc., 1999, 121, 2762–2769 CrossRef CAS; (b) M. R. Boyd, Y. F. Hallock, J. H. Cardellina, K. P. Manfredi, J. W. Blunt, J. B. McMahon, R. W. Buckheit, G. Bringmann and M. Schaeffer, J. Med. Chem., 1994, 37, 1740–1745 CrossRef CAS; (c) C. F. Nising, U. K. Schmid, M. Nieger and S. Bräse, J. Org. Chem., 2004, 69, 6830–6833 CrossRef CAS PubMed; (d) C. A. Mulrooney, B. J. Morgan, X. Li and M. C. Kozlowski, J. Org. Chem., 2010, 75, 16–29 CrossRef CAS PubMed; (e) Z. Li, Y. Gao, Y. Tang, M. Dai, G. Wang, Z. Wang and Z. Yang, Org. Lett., 2008, 10, 3017–3020 CrossRef CAS PubMed.
- (a) I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene-Based Materials, John Wiley & Sons, Chichester, U.K., 2009 Search PubMed; (b) A. Mori and A. Sugie, Bull. Chem. Soc. Jpn., 2008, 81, 548–561 CrossRef CAS; (c) F. Zhang and P. Bäuerle, J. Am. Chem. Soc., 2007, 129, 3090–3091 CrossRef CAS PubMed; (d) M. Takahashi, K. Masui, H. Sekiguchi, N. Kobayashi, A. Mori, M. Funahashi and N. Tamaoki, J. Am. Chem. Soc., 2006, 128, 10930–10933 CrossRef CAS PubMed; (e) E. J. Dell, B. Capozzi, K. H. DuBay, T. C. Berkelbach, J. R. Moreno, D. R. Reichman, L. Venkataraman and L. M. Campos, J. Am. Chem. Soc., 2013, 135, 11724–11727 CrossRef CAS PubMed; (f) K. W. R. de França, M. Navarro, É. Léonel, M. Durandetti and J.-Y. Nédélec, J. Org. Chem., 2002, 67, 1838–1842 CrossRef PubMed.
- (a) D. Kotowski, S. Luzzati, G. Scavia, M. Cavazzini, A. Bossi, M. Catellani and E. Kozma, Dyes Pigm., 2015, 120, 57–64 CrossRef CAS PubMed; (b) Y. Lin, Z.-G. Zhang, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2013, 1, 5128–5135 RSC; (c) J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed; (d) A. G. Macedo, D. C. Silva, N. A. D. Yamamoto, L. Micaroni, R. M. Q. Mello and L. S. Roman, Synth. Met., 2013, 170, 63–68 CrossRef CAS PubMed; (e) Y. Chen, Z. Du, W. Chen, L. Han, Q. Liu, M. Sun and R. Yang, Synth. Met., 2014, 187, 24–29 CrossRef CAS PubMed.
- (a) J. Yu, T.-L. Shen, W.-H. Weng, Y.-C. Huang, C.-I. Huang, W.-F. Su, S.-P. Rwei, K.-C. Ho and L. Wang, Adv. Energy Mater., 2012, 2, 245–252 CrossRef CAS PubMed; (b) U. Mehmood, I. A. Hussein, M. Daud, S. Ahmed and K. Harrabi, Dyes Pigm., 2015, 118, 152–158 CrossRef CAS PubMed; (c) X. Ren, Q. Feng, G. Zhou, C.-H. Huang and Z.-S. Wang, J. Phys. Chem. C, 2010, 114, 7190–7195 CrossRef CAS; (d) T. Duan, K. Fan, C. Zhong, T. Peng, J. Qin and X. Chen, RSC Adv., 2012, 2, 7081–7086 RSC; (e) W. Cao, M. Fang, Z. Chai, H. Xu, T. Duan, Z. Li, X. Chen, J. Qin and H. Han, RSC Adv., 2015, 5, 32967–32975 RSC.
- (a) Y. Ie, K. Nishida, M. Karakawa, H. Tada, A. Asano, A. Saeki, S. Seki and Y. Aso, Chem.–Eur. J., 2011, 17, 4750–4758 CrossRef CAS PubMed; (b) M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. J. Fréchet, Chem. Mater., 2013, 25, 4088–4096 CrossRef CAS; (c) T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed; (d) Y. Shu, A. Mikosch, K. N. Winzenberg, P. Kemppinen, C. D. Easton, A. Bilic, C. M. Forsyth, C. J. Dunn, T. B. Singh and G. E. Collis, J. Mater. Chem. C, 2014, 2, 3895–3899 RSC; (e) Z. Cai, H. Luo, P. Qi, J. Wang, G. Zhang, Z. Liu and D. Zhang, Macromolecules, 2014, 47, 2899–2906 CrossRef CAS.
- (a) K. Lee and P. H. Lee, Tetrahedron Lett., 2008, 49, 4302–4305 CrossRef CAS PubMed; (b) J. Hashim, T. N. Glasnov, J. M. Kremsner and C. O. Kappe, J. Org. Chem., 2006, 71, 1707–1710 CrossRef CAS PubMed; (c) Y. Miyake, M. Wu, M. J. Rahman, Y. Kuwatani and M. Iyoda, J. Org. Chem., 2006, 71, 6110–6117 CrossRef CAS PubMed; (d) X. Xu, D. Cheng and W. Pei, J. Org. Chem., 2006, 71, 6637–6639 CrossRef CAS PubMed; (e) P. H. Lee, D. Seomoon and K. Lee, Org. Lett., 2005, 7, 343–345 CrossRef CAS PubMed; (f) R. Satapathy, Y.-H. Wu and H.-C. Lin, Org. Lett., 2012, 14, 2564–2567 CrossRef CAS PubMed.
- (a) C. Gonzalez-Arellano, A. Corma, M. Iglesias and F. Sanchez, Chem. Commun., 2005, 1990–1992 RSC; (b) G. Cheng and M. Luo, Eur. J. Org. Chem., 2011, 2519–2523 CrossRef CAS PubMed; (c) P. Puthiaraj, P. Suresh and K. Pitchumani, Green Chem., 2014, 16, 2865–2875 CAS; (d) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788–13789 CrossRef CAS PubMed; (e) A. S. Demir, Ö. Reis and M. Emrullahoglu, J. Org. Chem., 2003, 68, 10130–10134 CrossRef CAS PubMed; (f) F. A. Arroyave, C. A. Richard and J. R. Reynolds, Org. Lett., 2012, 14, 6138–6141 CrossRef CAS PubMed.
- (a) C.-J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed; (b) S. Venkatraman, T. Huang and C.-J. Li, Adv. Synth. Catal., 2002, 344, 399–405 CrossRef CAS; (c) C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 5, 68–82 RSC; (d) B. Rao, W. Zhang, L. Hu and M. Luo, Green Chem., 2012, 14, 3436–3440 RSC; (e) B. Karimi, H. Behzadnia and H. Vali, ChemCatChem, 2014, 6, 745–748 CrossRef CAS PubMed; (f) H. Li, W. Chai, F. Zhang and J. Chen, Green Chem., 2007, 9, 1223–1228 RSC; (g) A. Kamal, V. Srinivasulu, B. N. Seshadri, N. Markandeya, A. Alarifib and N. Shankaraiah, Green Chem., 2012, 14, 2513–2522 RSC; (h) A. Monopoli, V. Calò, F. Ciminale, P. Cotugno, C. Angelici, N. Cioffi and A. Nacci, J. Org. Chem., 2010, 75, 3908–3911 CrossRef CAS PubMed.
- (a) Q. Wang, M. Wakioka and F. Ozawa, Macromol. Rapid Commun., 2012, 33, 1203–1207 CrossRef CAS PubMed; (b) M. Sévignon, J. Papillon, E. Schulz and M. Lemaire, Tetrahedron Lett., 1999, 40, 5873–5876 CrossRef.
- (a) K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS; (b) B. J. Campo, D. Bevk, J. Kesters, J. Gilot, H. J. Bolink, J. Zhao, J.-C. Bolsée, W. D. Oosterbaan, S. Bertho, J. D'Haen, J. Manca, L. Lutsen, G. van Assche, W. Maes, R. A. J. Janssen and D. Vanderzande, Org. Electron., 2013, 14, 523–534 CrossRef CAS PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- A. A. El-Shehawy, N. I. Abdo, A. A. El-Barbary and J.-S. Lee, Eur. J. Org. Chem., 2011, 4841–4852 CAS.
- M. J. Burns, I. J. S. Fairlamb, A. R. Kapdi, P. Sehnal and R. J. K. Taylor, Org. Lett., 2007, 9, 5397–5400 CrossRef CAS PubMed.
- K. Kawabata, M. Takeguchi and H. Goto, Macromolecules, 2013, 46, 2078–2091 CrossRef CAS.
- T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Tetrahedron, 2009, 65, 10797–10815 CrossRef CAS PubMed.
- J. C. Speros, H. Martinez, B. D. Paulsen, S. P. White, A. D. Bonifas, P. C. Goff, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2013, 46, 5184–5194 CrossRef CAS.
- B. Liu, W.-L. Yu, Y.-H. Lai and W. Huang, Macromolecules, 2000, 33, 8945–8952 CrossRef CAS.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- S. S. Gunathilake, H. D. Magurudeniya, P. Huang, H. Nguyen, E. A. Rainbolt, M. C. Stefan and M. C. Biewer, Polym. Chem., 2013, 4, 5216–5219 RSC.
- J. A. Letizia, S. Cronin, R. P. Ortiz, A. Facchetti, M. A. Ratner and T. J. Marks, Chem.–Eur. J., 2010, 16, 1911–1928 CrossRef CAS PubMed.
- C. Amatore, C. Cammoun and A. Jutand, Eur. J. Org. Chem., 2008, 4567–4570 CrossRef CAS PubMed.
- N.-N. Li, Y.-L. Zhang, S. Mao, Y.-R. Gao, D.-D. Guo and Y.-Q. Wang, Org. Lett., 2014, 16, 2732–2735 CrossRef CAS PubMed.
- J. Leroy, J. Levin, S. Sergeyev and Y. Geerts, Chem. Lett., 2006, 166–167 CrossRef CAS.
- N. I. Abdo, J. Ku, A. A. El-Shehawy, H.-S. Shim, J.-K. Min, A. A. El-Barbary, Y. H. Jang and J.-S. Lee, J. Mater. Chem. A, 2013, 1, 10306–10317 CAS.
- A. C. Jahnke, J. Proppe, M. Spulber, C. G. Palivan, C. Herrmann and O. S. Wenger, J. Phys. Chem. A, 2014, 118, 11293–11303 CrossRef CAS PubMed.
- G. Cheng and M. Luo, Eur. J. Org. Chem., 2011, 2519–2523 CrossRef CAS PubMed.
- B. R. Park, K. H. Kim, T. H. Kim and J. N. Kim, Tetrahedron Lett., 2011, 52, 4405–4407 CrossRef CAS PubMed.
- S. Zhang, D. Zhang and L. S. Liebeskind, J. Org. Chem., 1997, 62, 2312–2313 CrossRef CAS.
- L. Zhang, A. Wang, J. T. Miller, X. Liu, X. Yang, W. Wang, L. Li, Y. Huang, C.-Y. Mou and T. Zhang, ACS Catal., 2014, 4, 1546–1553 CrossRef CAS.
- F. Damkaci, E. Altay, M. Waldron, M. A. Knopp, D. Snow and N. Massaro, Tetrahedron Lett., 2014, 55, 690–693 CrossRef CAS PubMed.
- M. Boiocchi, M. Bonizzoni, L. Fabbrizzi, G. Piovani and A. Taglietti, Angew. Chem., Int. Ed., 2004, 43, 3847–3852 CrossRef CAS PubMed.
- N. Kirai and Y. Yamamoto, Eur. J. Org. Chem., 2009, 1864–1867 CrossRef CAS PubMed.
- G. Cahiez, C. Chaboche, F. Mahuteau-Betzer and M. Ahr, Org. Lett., 2005, 7, 1943–1946 CrossRef CAS PubMed.
- K. Mitsudo, T. Shiraga, D. Kagen, D. Shi, J. Y. Becker and H. Tanaka, Tetrahedron, 2009, 65, 8384–8388 CrossRef CAS PubMed.
- M. Zeng, Y. Du, C. Qi, S. Zuo, X. Li, L. Shao and X.-M. Zhang, Green Chem., 2011, 13, 350–356 RSC.
- C. Qi, X. Sun, C. Lu, J. Yang, Y. Du, H. Wu and X.-M. Zhang, J. Org. Chem., 2009, 694, 2912–2916 CrossRef CAS PubMed.
- R. B. Nasir Baig and R. S. Varma, Green Chem., 2013, 15, 398–417 RSC.
- C. Amatore, C. Cammoun and A. Jutand, Eur. J. Org. Chem., 2008, 4567–4570 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra (1H and 13C) for compounds 1, 2, 4. See DOI: 10.1039/c5ra13517f |
| This journal is © The Royal Society of Chemistry 2015 |