DOI:
10.1039/C5RA13426A
(Paper)
RSC Adv., 2015,
5, 76101-76106
Galvanic replacement-mediated synthesis of hollow Cu2O–Au nanocomposites and Au nanocages for catalytic and SERS applications†
Received
9th July 2015
, Accepted 1st September 2015
First published on 1st September 2015
Abstract
The galvanic replacement reaction (GRR) involves a corrosion process that is driven by the difference in the electrochemical potentials of two species. Here we demonstrate the synthesis of hollow Cu2O–Au nanocomposites via a GRR process between Cu2O and HAuCl4, and subsequent conversion of the hollow Cu2O–Au nanocomposites into Au nanocages that are actually assembled of ∼10 nm Au nanoparticles. It is interesting to find that Cu2O nanocubes produced from reductive solution chemistry are actually transformed from Cu(OH)2 nanowire precursors, and the Cu2O particle size is inversely proportional to the reaction temperature. A time-dependent TEM study of the GRR process between Cu2O and HAuCl4 indicates that this reaction involves evolution of an internal hollow core and surface precipitation of Au nanoparticles, which allows the formation of hollow Cu2O–Au nanocomposites. Comparing the properties of hollow Cu2O–Au nanocomposites and Au nanocages, it is determined that the hollow Cu2O–Au nanocomposites are more catalytically active in the reduction of 4-nitrophenol into 4-aminophenol in the presence of NaBH4, and Au nanocages are two orders of magnitude more sensitive in SERS detection of the target molecule, methylene blue. We believe the findings in this work may render a better understanding of the preparation and GRR process of Cu2O nanomaterials.
Introduction
The galvanic replacement reaction (GRR) has received more and more attention in preparing metal and metal oxide (sulfide) nanoparticles with controllable size, shape and properties,1–5 where a corrosion process that is driven by the difference in the electrochemical potentials of two species is involved. The GRR method allows selective deposition onto oxidizable structures, without the need of an external voltage and a reducing agent. Based on the chemical nature of the sacrificing template, GRR can lead to metal/metal oxide heterojunctions, metal nanoparticles, and even oxide/oxide nanostructures.4,6–10 In recent years, many sacrificing templates have been found to be suitable in the GRR, such as Cu2O, Mn3O4, MnO, Ag. For instance, Lee, et al.11 reported a GRR-based Pt deposition on Mn3O4 NPs through reaction between Mn3O4 and PtCl42− complexes, and the obtained Pt/Mn3O4 showed excellent oxygen reduction reaction properties. They also used carbon-encapsulated MnO nanoparticles or a Mn3O4-layer-coated interior surface of the hollow silica nanosphere as a reaction template for the selective decoration of the external surfaces with catalytic nanocrystals of various noble metals, including Pt, Pd, Rh, and Ir.10,12 Ag nanoparticles have been widely used as sacrificing templates to fabricate various metal and metal alloy nanostructures,13–21 which can be applied in catalysis, imaging, and therapy, etc. Cu2O has been widely used in GRR to produce functional metal nanoparticles and metal/oxide hybrid nanocomposites.22 Liu, et al.23 reported an in situ growth of Au NPs on the surfaces of Cu2O nanocubes. Rubinstein and co-workers systematically studied the pH-dependent galvanic replacement of supported and colloidal Cu2O nanocrystals with gold and palladium.2 In our previous work,6 self-supported Pt nanoclusters consisting of 2–3 nm NPs were synthesized via a GRR process between Cu2O and PtCl42− and we pointed out the importance of solution acidity for the GRR to proceed, where Cu2O was transformed into Cu2+ ions and surface-clean Pt electrocatalysts can be achieved. Ag nanosheet or nanoparticle assemblies could be prepared via reaction between Ag+ ions and Cu2O substrates, which show promise as surface-enhanced Raman scattering (SERS) substrates.7,24
Herein, we demonstrate the fabrication of hollow Cu2O–Au nanocomposites by careful control over the GRR process between Cu2O and HAuCl4, and then conversion of the as-prepared hollow Cu2O–Au nanocomposites into Au nanocages by an acid etching process (Scheme 1). The evolution of Cu2O nanocubes from Cu(OH)2 nanowire precursors has been found out, and the GRR process between Cu2O and HAuCl4 has been investigated by time-dependent TEM study. It is determined that the hollow Cu2O–Au nanocomposites and Au nanocages show different properties in catalytic and SERS applications. We believe this work may open new avenues for the synthesis of metal/oxide and metal nanostructures via GRR process for various applications.
 |
| | Scheme 1 Schematic illustration of the synthesis of hollow Cu2O–Au nanocomposites and Au nanocages through a galvanic replacement reaction (GRR) process between Cu2O and HAuCl4. | |
Experimental section
Synthesis of Cu2O nanocubes
Cu2O nanocubes were prepared through a modified reductive solution chemistry route.25,26 In a typical procedure, 0.5 g of poly(ethylene glycol) (PEG, MW: 2000) was first dissolved in 10 mL of Cu(Ac)2 aqueous solution (0.1 mM). Once PEG was completely dissolved, 50 μL of NaOH solution (6.0 M) was added dropwise. Upon addition, the solution immediately changed to blue color, indicating the formation of Cu(OH)2 precursors. After 10 min, 0.2 mL of ascorbic acid (AA) solution (1 M) was added dropwise to the solution, where the solution slowly turned into orange color. The products were collected by centrifugation after a reaction time of 1 h by repeatedly rinsing with DI water and ethanol in order to minimize the surface adsorbed PEG molecules. Here, the experiments were carried out at room temperature or in ice-water bath to study the effect of temperature in the growth of Cu2O nanocubes.
Synthesis of hollow Cu2O–Au nanocomposites and Au nanocages
Synthesis of hollow Cu2O–Au nanocomposites and Au nanocages was achieved through a galvanic replacement process between Cu2O and HAuCl4. For the synthesis of hollow Cu2O–Au nanocomposites, 40 mg of as-synthesized Cu2O nanocubes were ultrasonically re-dispersed in deionized water, and then 40 mL of HAuCl4 solution (1 mM) was added under magnetic stirring. The reaction was maintained for 5 min before the hollow Cu2O–Au nanocomposites were collected by centrifugation and rinsing with water and ethanol. For the synthesis of Au nanocages, 60 mL of HAuCl4 solution (1 mM) were used to prepare Cu2O–Au nanocomposites with more Au loading. Then, the as-prepared hollow Cu2O–Au nanocomposites were re-dispersed into 30 mL deionized water, followed by adding 10 mL of HCl (0.01 M) to dissolve the Cu2O. Stirring was not applied in order not to destroy the nanocage framework. The products were collected by centrifugation and rinsing with water and ethanol.
Catalytic reduction of 4-nitrophenol
The reduction of 4-nitrophenol was carried out in the presence of NaBH4 in a quartz cuvette and monitored by UV-Vis spectroscopy at room temperature.27 A total 60 μL of 4-nitrophenol aqueous solution (10 mM) was mixed with 0.16 mL of fresh NaBH4 (0.1 M) solution. Subsequently, 0.1 mL of hollow Cu2O–Au nanocomposites or Au nanocages dispersion (catalyst content: 1 mg mL−1) was added, and the mixture solution was quickly subjected to UV-Vis measurements to record the change in absorbance at a time interval of 1 min.
SERS measurements
Methylene blue (MB) dye was used as a Raman probe for the SERS sensitivity of the hollow Cu2O–Au nanocomposites and Au nanocages. Typically, aqueous dispersion of hollow Cu2O–Au nanocomposites or Au nanocages was injected into MB aqueous solution with variable concentrations ranging from 1.0 × 10−5 M to 1.0 × 10−9 M. After 1 h, the resulting mixture solution was centrifuged and re-dispersed on glass substrate and dried in air before the surface-enhanced Raman scattering (SERS) responses were determined.
Characterization
Powder X-ray diffraction (XRD) patterns of the samples were obtained on an XRD-6000 (Shimadzu) using a Cu Kα source (λ = 0.154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 056 nm). Transmission electron microscope (TEM, Tecnai G2 F20) and scanning electron microscope (SEM, FEI Quanta 200F) were applied to observe the particle size and surface morphology. The SERS spectra were recorded on a Renishaw In Via micro Raman spectroscopy system, using the TE air-cooled 576 × 400 CCD array in a confocal Raman system (wavelength: 633 nm).
056 nm). Transmission electron microscope (TEM, Tecnai G2 F20) and scanning electron microscope (SEM, FEI Quanta 200F) were applied to observe the particle size and surface morphology. The SERS spectra were recorded on a Renishaw In Via micro Raman spectroscopy system, using the TE air-cooled 576 × 400 CCD array in a confocal Raman system (wavelength: 633 nm).
Results and discussion
Preparation of Cu2O through the reductive chemistry route mainly involves two reactions: precipitation of Cu2+ ions with OH− ions into Cu(OH)2 precursor, and reduction of Cu(OH)2 into Cu2O by ascorbic acid (AA). In our previous work, we have demonstrated that different surfactants applied in the reaction system could lead to Cu2O nanoparticles with various nanostructures.25 Here, we investigated the reaction process by TEM study of the Cu(OH)2 precursor and Cu2O product, and found that the reaction temperature could also manipulate the Cu2O particle size. As shown in Fig. 1a and b, the blue Cu(OH)2 precursors obtained at room temperature and in ice-water bath are similar nanowires that are about 5–10 nm in diameter. Interestingly, these Cu(OH)2 nanowires are subsequently transformed into Cu2O nanocubes during the reductive process (Fig. 1c and d). Of note is that here we found that a lower temperature condition leads to Cu2O nanocubes with larger size: 40–60 nm at room temperature (Fig. 1c) vs. 100–120 nm in ice-water bath condition (Fig. 1d). This inverse relationship between reaction temperature and particle size might be interpreted from the nucleation and growth of Cu2O from Cu(OH)2 through the interfacial reduction. At a relatively high temperature, reaction between Cu(OH)2 and AA follows a rapid process, leading to more crystal nuclei and thus Cu2O nanoparticles with smaller size during the subsequent growth stage. While, at low temperature, a slow reaction process between Cu(OH)2 and AA produces limited nuclei, and Cu2O can grow into larger nanoparticles. Actually, higher temperature leading to Cu2O nanoparticles with smaller size has been seen in the growth of Cu2O@CeO2 core@shell nanocubes.28 However, the evolution of Cu2O nanocubes from Cu(OH)2 nanowires has not been reported previously and the underlying mechanism remains to be explored.
 |
| | Fig. 1 TEM images of Cu(OH)2 nanowire precursors prepared at room temperature (a) and in ice-water bath (b), and Cu2O nanocubes prepared at room temperature (c) and in ice-water bath (d). | |
Galvanic replacement reaction (GRR) between Cu2O and HAuCl4 follows the reaction,
| | |
3Cu2O + 6H+ + 2AuCl4− → 2Au + 6Cu2+ + 8Cl− + 3H2O
| (1) |
and this reaction follows a very rapid process, as the orange color of Cu
2O nanocubes will be immediately changed into dark brown upon interaction with HAuCl
4. And we found that the structure of Cu
2O nanocubes that are 40–60 nm in size (
Fig. 1 c) can be easily destroyed, and only scattered Au nanoparticles can be obtained (see Fig. S1 in ESI
†). Therefore, here we used the Cu
2O nanocubes that are 100–120 nm in size (
Fig. 1d) to study the galvanic replacement process between Cu
2O and HAuCl
4 (
Fig. 2), and this reaction involves evolution of an internal hollow core and surface precipitation of Au nanoparticles. It can be seen that the inside part of the Cu
2O nanocubes will be firstly etched by a mechanism analogous to pinhole corrosion, and the pinholes serve as transport paths during the dissolution of Cu
2O nanocubes. With the elongation of reaction time, hole expansion and Au precipitation result in hollow Cu
2O–Au nanocomposites. However, given long enough reaction time and adequate HAuCl
4 source, the Cu
2O will be fully dissolved and the nanocube framework will be eventually collapsed, resulting in only dispersed Au nanoparticles (see Fig. S2 in ESI
†). It has been reported that reaction between HAuCl
4 and Cu
2O might lead to CuO,
22 but we found Cu
2O would be transformed into soluble Cu
2+ species in acidic environment.
6
 |
| | Fig. 2 Time-dependent TEM study of the galvanic replacement process between Cu2O and HAuCl4, (a) 30 s, (b) 1 min, (c) 2 min, and (d) 5 min. Scale bar: 50 nm. | |
Before the Cu2O is completely dissolved, it is easy to get hollow Cu2O–Au nanocomposites (Fig. 3a and b), which consist of 5–10 nm Au nanoparticles and internally hollow Cu2O. From the HR-TEM image in Fig. 3b, one can clearly see that Au nanoparticles grown along the (111) plane are decorated on the Cu2O surface. As mentioned above, without experimental control, the Cu2O nanocube framework will be eventually collapsed and only dispersed Au nanoparticles are produced. Here, we used an acid etching technique to obtain well-defined Au nanocages (Fig. 3c and d), where more HAuCl4 is used to increase the Au loading. The obtained Cu2O–Au nanocomposites were then re-dispersed into water and subject to HCl etching to remove the Cu2O. It is interesting that the framework of the nanocubes is not destroyed and the obtained Au nanocages are comprised of an assembly of ∼10 nm Au nanoparticles, and the size of the nanocage is similar to that of the Cu2O nanocubes. HR-TEM image of the Au nanocages shows that these highly crystallized Au nanoparticles are also grown mainly along the (111) plane. The hollow Cu2O–Au and Au nanocage structures can also be confirmed from the SEM studies (see Fig. S3 in ESI†). Therefore, by careful control of the galvanic replacement process, we are able to get hollow Cu2O–Au nanocomposites and Au nanocages.
 |
| | Fig. 3 TEM and HR-TEM images of Cu2O–Au nanocomposites (a and b) and Au nanocages (c and d) prepared from a galvanic replacement process between Cu2O and HAuCl4. | |
The successful formation of Cu2O–Au nanocomposites and subsequent transformation into Au nanocages can be confirmed by the X-ray diffraction (XRD) patterns in Fig. 4. The diffraction peaks appeared at 2θ = ∼29.6, 36.5, 42.4, 52.6, 61.5, 73.7 and 77.6° (labeled with *) in the Cu2O nanocubes can be well indexed to the (110), (111), (200), (211), (220), (311), and (222) crystal planes of the Cu2O crystals in cubic phase (PDF No. 65-3288), indicating the production of pure Cu2O samples, without any CuO or Cu(OH)2 impurities. Besides the diffraction peaks of Cu2O in the Cu2O–Au nanocomposites, one can also see diffraction peaks at 2θ = ∼38.2, 44.6, 64.7, and 77.5°, corresponding to the (111), (200), (220), (311) crystal planes (labeled with o) of face-center-cubic (fcc) Au crystals (PDF No. 01-1172). After acid etching, the Cu2O can be completely removed and only diffraction peaks of Au can be seen in the as-prepared Au nanocages.
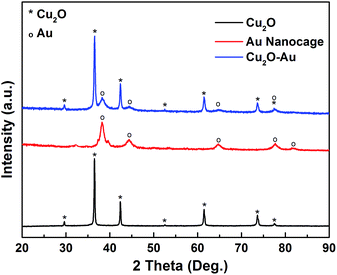 |
| | Fig. 4 X-ray diffraction (XRD) patterns of the as-prepared Cu2O nanocubes, Cu2O–Au nanocomposites and Au nanocages. | |
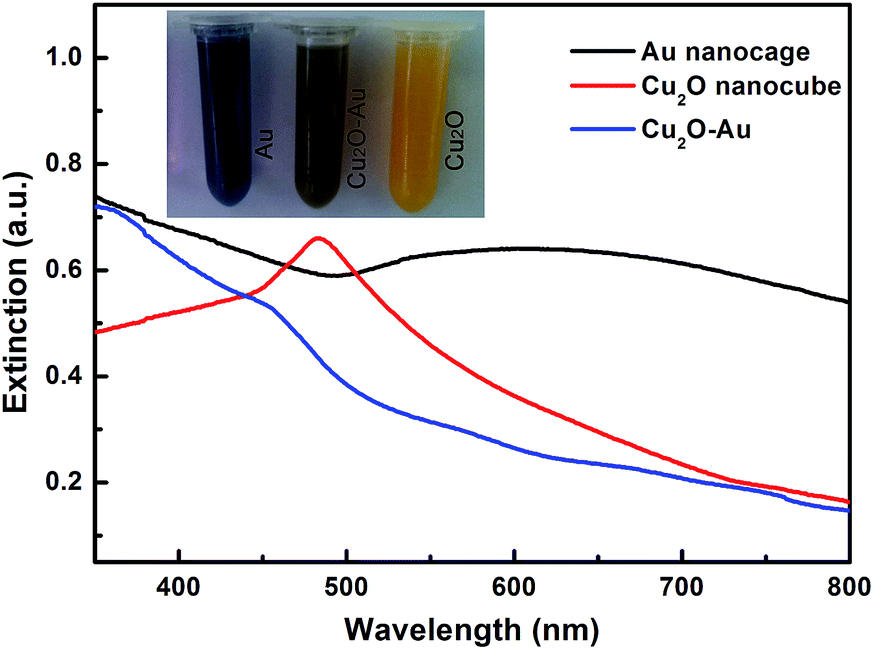
From the optical images inset in Fig. 5, one can see that the as-prepared Cu2O nanocubes are orange in color, and the hollow Cu2O–Au nanocomposites become dark brown. While, the Au nanocages, which are about 100 nm in size and assembled by tiny nanoparticles, display darkish blue color. Extinction spectra of these samples dispersed in water are also determined. The Cu2O nanocubes that are 100–120 nm in size show an absorption peak at about 485 nm, which agrees well with the previous reports.26 The Au nanocages that are ∼100 nm in size have a very broad absorption band centered at ∼610 nm. Different from reported Au nanocages that usually show strong absorption at ∼800 nm,29 here we think the red shift of the absorption peak can be interpreted by the fact that the prepared Au nanocages are actually assembled by ∼10 nm nanoparticles (Fig. 3c). While, it is interesting to see that the extinction spectrum of the hollow Cu2O–Au nanocomposites differs a lot from those of Cu2O nanocubes and Au nanocages, where no distinct absorption peak can be detected. We think this can be understood through the TEM studies (Fig. 2 and 3), where very limited Cu2O remains in the Cu2O–Au nanocomposites during the galvanic replacement process. Moreover, aggregation of Au nanoparticles on the remained Cu2O frameworks to give the new hollow Cu2O–Au nanocomposites may also cause this phenomenon. An absorption shoulder at ∼560 nm can well reflect the size of Au nanoparticles supported on the hollow Cu2O edges.3 A dynamic light scattering (DLS) study of the Cu2O–Au nanocomposites and Au nanocages shows that they are quite stable in solution, and the size distribution agrees well with the TEM results (see Fig. S4 in ESI†).
 |
| | Fig. 5 Extinction spectra of Cu2O nanocubes, hollow Cu2O–Au nanocomposites, and Au nanocages. Inset shows the optical images of these samples dispersed in water. | |
Au nanoparticles are promising catalysts for various chemical reactions and assemblies of Au nanoparticles are suitable for chemical detection based on surface enhanced Raman spectroscopy (SERS).30–33 Here, we tested the properties of the as-prepared hollow Cu2O–Au nanocomposites and Au nanocages in catalytic reduction of 4-nitrophenol in the presence of NaBH4 and SERS detection of methylene blue (MB). The reduction of 4-nitrophenol into 4-aminophenol can be monitored by the absorption peak at about 405 nm in the UV-Vis spectroscopy (see Fig. S5 in ESI†). As shown in Fig. 6, the as-prepared 100–120 nm Cu2O nanocubes almost have no catalytic effect to the reduction of 4-nitrophenol. The hollow Cu2O–Au nanocomposites are highly efficient for this reaction, as the 4-nitrophenol can be fully converted into 4-aminophenol at a time scale of 6 min. Surprisingly, the Au nanocages are less effective in catalyzing this reaction, since only ∼55% of 4-nitrophenol are reacted after 8 min. Aggregation of Au nanoparticles usually occurs during the catalytic process in solution, which will decrease the catalytic activity and cycling performance. Therefore, many works have been focused on the fabrication of Au nanoparticles on various carriers, which can effectively reduce the aggregation and thus maintain the catalytic activity.34–36 Here, we think the Au nanoparticles supported on hollow Cu2O nanostructures are more active in catalyzing this reaction, and Au nanocages, which are essentially an assembly of Au nanoparticles, may suffer from particle aggregation through Ostwald ripening and reduction of surface area during the reaction process (see Fig. S6 in ESI†).
 |
| | Fig. 6 Catalytic performance of reduction of 4-nitrophenol into 4-aminophenol in the presence of NaBH4 using Cu2O nanocubes, hollow Cu2O–Au nanocomposites, and Au nanocages as catalysts. | |
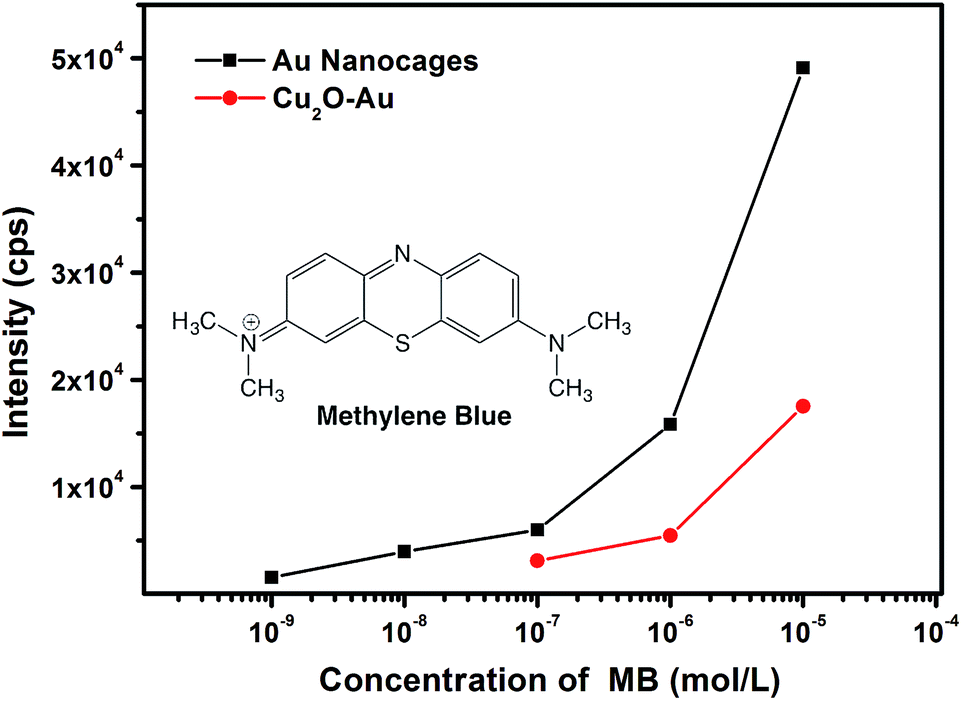
It is interesting to find that as compared with the catalysis study, hollow Cu2O–Au nanocomposites and Au nanocages have an inverse effect in SERS detection, where Au nanocages are more sensitive and promising as SERS platforms. With methylene blue (MB) as the target molecule, well-resolved Raman fingerprints can be distinguished on both hollow Cu2O–Au nanocomposites and Au nanocages (see Fig. S7 in ESI†). However, Au nanocages can be two orders of magnitude more sensitive in SERS detection. As shown in Fig. 7, with the Raman intensity of the ν(C–C) ring stretching at 1618 cm−1 of MB as a reference, a detection limit of 10−7 M was found on hollow Cu2O–Au nanocomposites, which can be as low as 10−9 M on Au nanocages. Moreover, at the same concentration of MB, the Raman intensity is much stronger on Au nanocages, an indication that Au nanocages are more suitable as a SERS platform for chemical detection. This can be explained from the SERS image of the two samples, where more hot spots with stronger Raman intensity can be detected on an aggregation of Au nanocages (see Fig. S8 in ESI†). This clearly shows that the Au nanoparticle assemblies in the nanocages can generate more hot spots for SERS detection, since the gaps and junctions between two adjacent Au nanoparticles are usually favorable to create much enhanced electromagnetic fields.37 In order to show the generality of Au nanocages for SERS detection, we have used another molecule, thiram, one kind of pesticides as an analyte. It was found that the detection limit of thiram could also reach 10−9 M on Au nanocages (see Fig. S9 in ESI†). SERS response in the NIR region has been of great interest, and it might be of great interest to test the SERS performances of the as-prepared materials if NIR laser sources are available.38
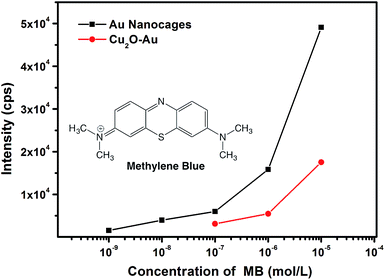 |
| | Fig. 7 Concentration-dependent Raman intensity of the ν(C–C) ring stretching at 1618 cm−1 of methylene blue (MB) using hollow Cu2O–Au nanocomposites and Au nanocages as SERS substrates. | |
Conclusions
In conclusion, we have demonstrated here the structure evolution of Cu2O nanocubes in reductive solution chemistry route and the synthesis of hollow Cu2O–Au nanocomposites and Au nanocages via a controlled galvanic replacement reaction (GRR) process. Cu2O nanocubes are transformed from Cu(OH)2 nanowire precursors, and the size of Cu2O nanocubes shows an inverse relationship with the reaction temperature. The GRR process between Cu2O and HAuCl4 follows a mechanism analogous to pinhole corrosion, allowing the formation of hollow Cu2O–Au nanocomposites. Au nanocages are then obtained by an acid etching treatment to remove the Cu2O of the hollow Cu2O–Au nanocomposites. The hollow Cu2O–Au nanocomposites, with Au nanoparticles well dispersed on the Cu2O framework, are promising catalyst for the reduction of 4-nitrophenol in the presence of NaBH4. On the contrary, Au nanocages, formed by an assembly of 10 nm Au nanoparticles, are more sensitive in chemical detection based on SERS technique, due to the generation of more hot spots by gaps and junctions between two adjacent Au nanoparticles. We believe the findings in this work may be intended to the synthesis of hollow nanostructures for various applications.
Acknowledgements
We thank the support from NSFC (No. 21471039, 21203045), Fundamental Research Funds for the Central Universities (Grant No. HIT. NSRIF. 2010065 and 2011017, PIRS of HIT A201502, and HIT.BRETIII. 201223), China Postdoctoral Science Foundation (2013M541394, 2014M560253 and 2014T70341), Natural Science Foundation of Heilongjiang Province (B2015001), Heilongjiang Postdoctoral Fund (LBH-Z14076), National Undergraduate Training Program for Innovation and Entrepreneurship, and Postdoctoral Scientific Research Developmental Fund of Heilongjiang Province (LBH-Q14062).
Notes and references
- E. Sutter, K. Jungjohann, S. Bliznakov, A. Courty, E. Maisonhaute, S. Tenney and P. Sutter, Nat. Commun., 2014, 5, 4946 CrossRef CAS PubMed.
- M. D. Susman, R. Popovitz-Biro, A. Vaskevich and I. Rubinstein, Small, 2015, 11, 3942–3953 CrossRef CAS PubMed.
- S. E. Skrabalak, J. Y. Chen, Y. G. Sun, X. M. Lu, L. Au, C. M. Cobley and Y. N. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS PubMed.
- M. H. Oh, T. Yu, S. H. Yu, B. Lim, K. T. Ko, M. G. Willinger, D. H. Seo, B. H. Kim, M. G. Cho, J. H. Park, K. Kang, Y. E. Sung, N. Pinna and T. Hyeon, Science, 2013, 340, 964–968 CrossRef CAS PubMed.
- E. Gonzalez, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377–1380 CrossRef CAS PubMed.
- Q. Li, P. Xu, B. Zhang, G. Wu, H. T. Zhao, E. G. Fu and H. L. Wang, Nanoscale, 2013, 5, 7397–7402 RSC.
- W. Jin, P. Xu, L. Xiong, Q. Jing, B. Zhang, K. Sun and X. J. Han, RSC Adv., 2014, 4, 53543–53546 RSC.
- B. E. Dixon, M. Rosenman, Y. N. Xia and S. J. Grannis, Stud. Health Tech. Informat., 2013, 192, 884–888 Search PubMed.
- W. Yao, F. L. Li, H. X. Li and J. P. Lang, J. Mater. Chem. A, 2015, 3, 4578–4585 CAS.
- D. G. Lee, S. M. Kim, H. Jeong, J. Kim and I. S. Lee, ACS Nano, 2014, 8, 4510–4521 CrossRef CAS PubMed.
- K. W. Kim, S. M. Kim, S. Choi, J. Kim and I. S. Lee, ACS Nano, 2012, 6, 5122–5129 CrossRef CAS PubMed.
- S. M. Kim, M. Jeon, K. W. Kim, J. Park and I. S. Lee, J. Am. Chem. Soc., 2013, 135, 15714–15717 CrossRef CAS PubMed.
- L. Au, X. M. Lu and Y. N. Xia, Adv. Mater., 2008, 20, 2517–2522 CrossRef CAS PubMed.
- V. Bansal, A. P. O'Mullane and S. K. Bhargava, Electrochem. Commun., 2009, 11, 1639–1642 CrossRef CAS PubMed.
- M. H. Cao, L. Zhou, X. Q. Xu, S. Cheng, J. L. Yao and L. J. Fan, J. Mater. Chem. A, 2013, 1, 8942–8949 CAS.
- J. Y. Chen, B. Wiley, J. McLellan, Y. J. Xiong, Z. Y. Li and Y. N. Xia, Nano Lett., 2005, 5, 2058–2062 CrossRef CAS PubMed.
- X. Y. Liu, A. Q. Wang, L. Li, T. Zhang, C. Y. Mou and J. F. Lee, Prog. Nat. Sci. Mat. Int., 2013, 23, 317–325 CrossRef PubMed.
- X. M. Lu, H. Y. Tuan, J. Y. Chen, Z. Y. Li, B. A. Korgel and Y. N. Xia, J. Am. Chem. Soc., 2007, 129, 1733–1742 CrossRef CAS PubMed.
- C. X. Wang, Y. Wang, L. Xu, X. D. Shi, X. W. Li, X. W. Xu, H. C. Sun, B. Yang and Q. Lin, Small, 2013, 9, 413–420 CrossRef CAS PubMed.
- Y. Yang, J. Y. Liu, Z. W. Fu and D. Qin, J. Am. Chem. Soc., 2014, 136, 8153–8156 CrossRef CAS PubMed.
- C. Zhang, Y. Zhang, D. K. Yao, Y. N. Xia and L. H. V. Wang, Photons Plus Ultrasound: Imaging and Sensing 2012, 2012, vol. 8223 Search PubMed.
- M. L. Pang, Q. X. Wang and H. C. Zeng, Chem.–Eur. J., 2012, 18, 14605–14609 CrossRef CAS PubMed.
- X. W. Liu, F. Y. Wang, F. Zhen and J. R. Huang, RSC Adv., 2012, 2, 7647–7651 RSC.
- T. Gao, Y. Q. Wang, K. Wang, X. L. Zhang, J. N. Dui, G. M. Li, S. Y. Lou and S. M. Zhou, ACS Appl. Mater. Interfaces, 2013, 5, 7308–7314 CAS.
- Q. Li, P. Xu, B. Zhang, H. Tsai, S. J. Zheng, G. Wu and H. L. Wang, J. Phys. Chem. C, 2013, 117, 13872–13878 CAS.
- L. F. Gou and C. J. Murphy, J. Mater. Chem., 2004, 14, 735–738 RSC.
- F. H. Lin and R. A. Doong, J. Phys. Chem. C, 2011, 115, 6591–6598 CAS.
- X. Wang, D. P. Liu, J. Q. Li, J. M. Zhen and H. J. Zhang, NPG Asia Mater., 2015, 7, e158 CrossRef CAS PubMed.
- J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, Z. Y. Li, L. Au, H. Zhang, M. B. Kimmey, X. D. Li and Y. N. Xia, Nano Lett., 2005, 5, 473–477 CrossRef CAS PubMed.
- B. Zhang, B. T. Zhao, S. H. Huang, R. Y. Zhang, P. Xu and H. L. Wang, CrystEngComm, 2012, 14, 1542–1544 RSC.
- W. Y. Li, J. G. Liu and C. W. Yan, Carbon, 2013, 55, 313–320 CrossRef CAS PubMed.
- H. Zhu, M. L. Du, D. L. Yu, Y. Wang, L. N. Wang, M. L. Zou, M. Zhang and Y. Q. Fu, J. Mater. Chem. A, 2013, 1, 919–929 CAS.
- J. Zeng, Q. Zhang, J. Y. Chen and Y. N. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- J. Li, C. Y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- Q. M. Ji, J. P. Hill and K. Ariga, J. Mater. Chem. A, 2013, 1, 3600–3606 CAS.
- J. W. Xie, W. Y. Liu, M. R. MacEwan, P. C. Bridgman and Y. N. Xia, ACS Nano, 2014, 8, 1878–1885 CrossRef CAS PubMed.
- P. Xu, N. H. Mack, S. H. Jeon, S. K. Doorn, X. J. Han and H. L. Wang, Langmuir, 2010, 26, 8882–8886 CrossRef CAS PubMed.
- H. N. Xie, I. A. Larmour, Y. C. Chen, A. W. Wark, V. Tileli, D. W. McComb, K. Faulds and D. Graham, Nanoscale, 2013, 5, 765–771 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S9. See DOI: 10.1039/c5ra13426a |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.