
LTQ-Orbitrap-based strategy for traditional Chinese medicine targeted class discovery, identification and herbomics research: a case study on phenylethanoid glycosides in three different species of Herba Cistanches†
Abstract
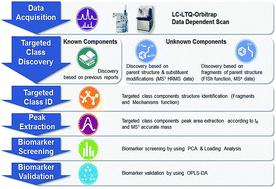
Traditional Chinese Medicine (TCM) usually contains one specific class of components that could represent its phytochemical properties and therapeutic effects. However, the unclear specific class constituents hinders the sufficient interpretation of its bioactivities. In this paper, an HPLC-LTQ-Orbitrap-based strategy focused on the TCM targeted class was developed. This strategy was successfully applied in the discovery and identification of phenylethanoid glycosides (PhGs) in Herba Cistanches, and herbomics research of Cistanche deserticola, C. sinensis, and C. tubulosa. A total of 69 PhGs, including 17 new PhGs, were rapidly discovered and characterized using multiple data-mining methods such as accurate parent mass search, parent mass modification search and fragment ion search, allowing a comprehensive revelation of PhGs in Cistanche species for the first time. Based on the peak areas of the 69 PhGs identified, multivariate statistical analysis such as PCA, loading analysis and OPLS-DA were employed in herbomics research to screen and validate the PhGs that could be utilized to discriminate these three Cistanche species. Eight PhGs were finally screened and chosen as chemical markers for the species discrimination. In conclusion, this new established strategy could be exemplary for future studies on the discovery and identification of important chemical constituents, and differentiation of genuine specie or genus from adulterants. Meanwhile, it may propose a novel idea for analyzing a specific class of active chemical constituents, and is promising for quality control and evaluation of TCMs.
 Please wait while we load your content...
Please wait while we load your content...

