
Rational design of nanoparticle/monomer interfaces: a combined computational and experimental study of in situ polymerization of silica based nanocomposites†‡
Abstract
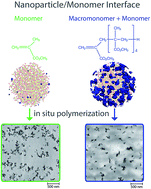
Interfaces between methylmethacrylate monomers, oligomers and silica nanoparticles (NPs) were explored by molecular dynamics simulations, infrared and solid state nuclear magnetic resonance spectroscopy. This knowledge allowed the control of the structure of the interfaces by employment of MMA macromonomers, and the design of an improved process for in situ polymerizations with a remarkable increase of NP dispersion.
 Please wait while we load your content...
Please wait while we load your content...

