
Electrocatalytic properties of iron chalcogenides as low-cost counter electrode materials for dye-sensitized solar cells†
Abstract
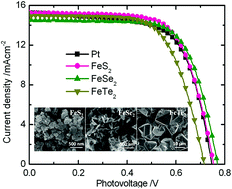
Developing cost-effective and highly electrocatalytic Pt-free counter electrode (CE) materials for triiodide reduction has become a major interest for dye-sensitized solar cells (DSCs). In a heterogeneous catalytic system, iron chalcogenides like FeS2 and FeSe2 have demonstrated excellent catalytic activity when serving as CE materials in DSCs. However, theoretical and experimental studies have yet to be conducted to investigate the catalytic activity of iron chalcogenides in energy conversion and storage devices under the same conditions. In this work, FeS2, FeSe2, and FeTe2 were selected as our research object to systematically investigate and compare the regulatory mechanisms of the changes in the catalytic activity of iron chalcogenides. Theoretical research reveals that the iodine adsorption and charge exchange of these three materials occurred efficiently in heterogeneous catalytic systems. Experimental results further show that these three materials exhibited excellent catalytic activities. The conversion efficiencies of the corresponding DSCs are comparable to those of the sputtered Pt CE. This study also provides a method to rationally screen cost-effective and highly efficient catalytic materials for electrocatalysis applications.
 Please wait while we load your content...
Please wait while we load your content...

