
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hole-transporting side-chain polystyrenes based on TCTA with tuned glass transition and optimized electronic properties†
Felix R. P.
Limberg
a,
Arunas
Miasojedovas
b,
Patrick
Pingel
a,
Felix
Reisbeck
a,
Silvia
Janietz
a,
Andrew P.
Monkman
b and
Hartmut
Krüger
*a
aFraunhofer Institute for Applied Polymer Research, Wissenschaftspark Golm, Geiselbergstr. 69, D-14476 Potsdam, Germany. E-mail: hartmut.krueger@iap.fraunhofer.de
bPhysics Department, University of Durham, South Road, Durham DH1 3LE, UK
First published on 30th September 2015
Abstract
The development of crosslinkable materials for the fabrication of solution processable OLEDs presents challenges, especially regarding the adjustment of the glass transition (Tg), which has a significant influence on crosslinking kinetics and device life-time. Crosslinkable hole transport materials based on poly(N,N-bis(4-(9H-carbazol-9-yl)phenyl)-4-vinylaniline) (poly-TCTA) with covalently attached plasticizers for Tg control and azide functionalities for azide-alkyne crosslinking are presented. These polymers have an optimal Tg of around 150 °C and show superior crosslinking performances and solution resistibilities. Incorporation of electron-pushing alkoxides to the pendant groups combines the Tg adjustment approach with a systematic tuning of the HOMO level from −5.7 to −5.3 eV. All presented polymers have good charge transport and injection properties and are ideal for applications in phosphorescent OLEDs due to their high triplet energies (>2.8 eV). The new crosslinkable poly-TCTA-based materials are applied as hole-transport layers (HTLs) in fully solution-processed OLEDs. An improvement of the device performance is demonstrated for OLEDs with additional crosslinked HTL.
Introduction
Modern organic light-emitting diodes (OLEDs) consist of several different functional materials and layers with well-defined thicknesses and morphologies. In order to realize such multi-layer structures, most OLEDs are still based on small molecules which often need to be processed under high-cost vacuum conditions. Therefore, their use is still limited to expensive high-resolution display applications, in, e.g., cell-phones and televisions, and to rare examples of expensive lighting applications.1,2 As an alternative, solution-based processes enable high throughput and low-cost fabrication of OLEDs on large size. However, solvent-processing of organic multi-layers is still difficult; e.g., controlling de-wetting, crystallization, or re-dissolving of deposited layers is a challenge. Orthogonal solvent strategies3 or stamping techniques4 can circumvent these problems but put severe constraints on the choice of compatible materials, number of layers, or solvents. Crosslinking techniques are a feasible alternative towards fully solution-processed multi-layer OLEDs and may enable low-cost lighting applications.5Crosslinking of organic layers generally requires exposure of the sample to UV light, to elevated temperatures, or to both in order to activate and promote the crosslinking reaction.5 Thereby, completion of the reaction can only be reached, if the sample is heated above the glass transition or melting temperature, at least once in a final curing step.6 Below the glass transition, the crosslinker groups are “frozen” in the glassy state and hence their reaction is restricted. For an economical and gentle processing, the glass transition temperature of a crosslinkable material should be as low as possible; however, a strong correlation between a high glass transition (Tg), morphological stability of organic layers, and long OLED lifetimes is apparent.7 In order to meet both processing and lifetime demands, we believe that a moderate glass transition, i.e., a moderate crosslinking temperature, between 150 °C and 170 °C would be ideal.
Recently, we introduced poly(N,N-bis(4-(9H-carbazol-9-yl)phenyl)-4-vinylaniline) (poly-TCTA) as host material in green phosphorescent OLEDs.8 In this polymer, a tris(4-(9H-carbazol-9-yl)phenyl)amine (TCTA) analogue is bound as pendant group to a polystyrene backbone. Polystyrene-based side chain functionalized polymers are known for excellent film formation and the morphological stability of such layers.9,10 Good hole-transporting properties and the high triplet energy (2.8 eV) of poly-TCTA led to good overall electroluminescent performances. Further device optimization might be possible by introducing a hole transport layer (HTL) between anode and the emission layer. The HTL material should possess a HOMO energy that compensates for the energy offset of the anode's work function (PEDOT:PSS ≈ 5.0 eV)11 and the polymer's HOMO-derived hole transport states (e.g., poly-TCTA, −5.6 eV) in order to improve charge carrier injection and to reduce the driving voltage. The HTL may also serve as an electron blocking layer.
Liaptsis and Meerholz introduced electron-pushing or -pulling groups to the aromatic core of a crosslinkable derivative of the hole-transporting small molecule 1-bis[4-[N,N-di(4-tolyl)amino]phenyl]cyclohexane (TAPC).12 This allowed for a systematic variation of the HOMO energy of these compounds and, in turn, led to tunability of the hole injection barrier in their OLED devices. In our work, we follow similar approaches to modify the chemical structure, electrooptical, and physical properties of poly-TCTA towards its applicability as crosslinked HTL.
Here, we present strategies to reduce the relatively high glass transition (247 °C) of side-chain poly-TCTA by either introducing plasticizers as co-monomers into the polymer chain or by attaching flexible substituents directly at the functional side-chain units. The glass transition (Tg) can be tuned in a broad range. The latter approach has been extended towards the use of electron pushing substituents which provides a systematical tunability of the HOMO level. The crosslinkable new HTL polymers are applied in OLED stacks.
Results and discussion
In general, the Tg of polymers can be reduced by a variety of commercially available plasticizers. However, their application in OLEDs is strongly limited, because they must meet the following criteria: (1) miscibility with the host; (2) chemical and optical inertness; (3) no diffusion into adjacent layers (therefore, a covalently bond plasticizer would be favorable); (4) no detrimental interference with charge transport or light emissive materials.T g control by covalently attached plasticizers
The first straight forward approach is the covalent attachment of a plasticizer as a co-monomer into the polymer (Scheme 1). The homo-polymer of 1-decyl-4-vinylbenzene (P) has a very deep Tg of about −65 °C, the lowest reported Tg of p-alkylstyrenes,13 which makes this compound a strong plasticizer. In addition, the plasticizer is not conjugated and is therefore expected to have no influence on the optical properties which are determined by the TCTA units.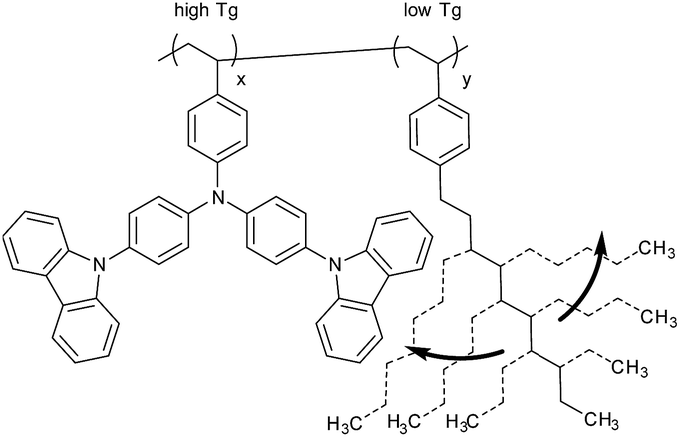 | ||
| Scheme 1 Copolymeric Tg reduction approach using 1-decyl-4-vinylbenzene as plasticizer (P). | ||
1-Decyl-4-vinylbenzene (P) was copolymerized with mono-TCTA (2a); the P feed ratios are 25 M% (12 wt%) for polymer 3a25, 50 M% (29 wt%) for 3a50, and 75 M% (55 wt%) for 3a75 (Scheme 2). With increasing amount of plasticizer the number-average molecular weight (Mn) is only slightly decreased, whereas the weight-average molecular weight (Mw) decreases significantly accompanied by lower polydispersities (PDI) of the copolymers. This can be explained by the reduced average monomer weight at higher plasticizer content. The amount of P in the copolymers, determined with 1H-NMR, is in good agreement with the feed ratios. Selected properties of the prepared polymer samples are summarized in Tables 1 and 2.
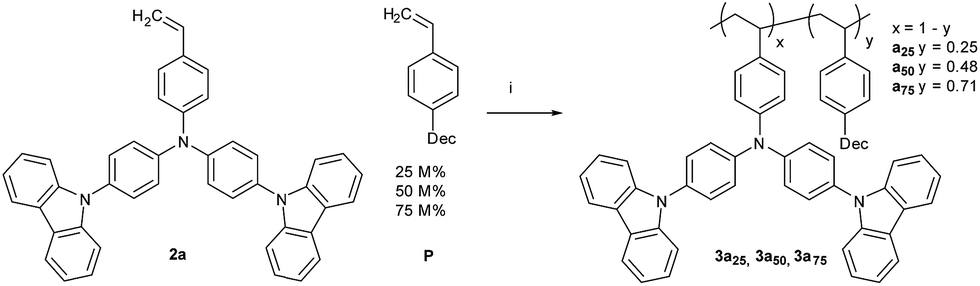 | ||
| Scheme 2 Synthetic route towards Tg adapted poly-TCTA copolymers with 1-decyl-4-vinylbenzene (P) as comonomer. | ||
| Polymer | Yield [%] | M n a [103 g mol−1] | M w a [103 g mol−1] | PDI | DPnb | T g c [°C] |
|---|---|---|---|---|---|---|
| a Determined with size exclusion chromatography (SEC) using polystyrene standards. b Degree of polymerization: Mn × Mmonomer − 1. c Glass transition determined with differential scanning calorimetry (DSC), in brackets: self-crosslinked. | ||||||
| 3a | 92 | 27.6 | 112 | 4.04 | 46 | 247 |
| 3a25 | 76 | 33.3 | 111 | 3.34 | 65 | 203 |
| 3a50 | 67 | 29.4 | 70.9 | 2.41 | 70 | 162 |
| 3a75 | 58 | 20.8 | 47.3 | 2.27 | 62 | 87.2 |
| 3aAZ15 | 81 | 20.6 | 60.8 | 2.95 | 39 | −(251) |
| 3a43AZ15 | 61 | 21.8 | 46.6 | 2.14 | 57 | 172 (173) |
| 3b | 56 | 9.05 | 22.7 | 2.51 | 14 | 220 |
| 3c | 81 | 18.5 | 73.7 | 3.98 | 22 | 144 |
| 3d | 60 | 20.9 | 51.1 | 2.44 | 23 | 146 |
| 3e | 72 | 29.2 | 60.9 | 2.09 | 38 | 184 |
| 3f | 64 | 24.3 | 57.8 | 2.38 | 27 | 150 |
| 3fAZ15 | 71 | 16.9 | 30.3 | 1.79 | 22 | 144 (150) |
| Polymer | Absorption max.a [eV] | Fluorescence max.b [eV] | Stokes shift [eV] | LUMOc [eV] | HOMOd [eV] | HOMOe [eV] | E G f [eV] | E T g [eV] |
|---|---|---|---|---|---|---|---|---|
| a As solid film. b Photoluminescence spectra as solid film. c LUMO values were calculated by using the optical bandgap and the HOMO-values from PESA measurements. d Photoelectron spectroscopy in air (PESA). e Cyclovoltammetry peak potential against ferrocene standard. f Onset from UV/Vis-spectra. g For triplet energy levels the onsets of phosphorescence spectra were taken. | ||||||||
| 3a | 3.79 | 2.91 | 0.88 | −2.27 | −5.62 | −5.48 | 3.35 | 2.8 |
| 3a25 | 3.79 | 2.91 | 0.88 | −2.24 | −5.60 | −5.45 | 3.36 | 2.88 |
| 3a50 | 3.79 | 2.96 | 0.83 | −2.19 | −5.55 | −5.52 | 3.36 | 2.77 |
| 3a75 | 3.78 | 3.06 | 0.72 | −2.19 | −5.55 | −5.49 | 3.36 | 2.9 |
| 3b | 4.01 | 3.05 | 0.96 | −2.18 | −5.49 | −5.47 | 3.31 | 2.9 |
| 3c | 4.01 | 3.09 | 0.96 | −2.19 | −5.58 | −5.45 | 3.39 | 2.9 |
| 3d | 3.83 | 3.02 | 0.81 | −2.16 | −5.53 | −5.39 | 3.37 | 2.87 |
| 3e | 3.94 | 3.02 | 0.92 | −2.19 | −5.29 | −5.35 | 3.11 | 2.8 |
| 3f | 3.94 | 3.02 | 0.92 | −2.17 | −5.29 | −5.24 | 3.12 | 2.81 |
A strong Tg decrease was observed with increasing amounts of plasticizer P as expected. This decrease shows a linear dependence between the measured glass transition and the weight percentage of P (Fig. 1), wherein 3a50 (Tg = 162 °C) is in the desired Tg-range for OLED fabrication, while 3a25 (Tg = 203 °C) and 3a75 (Tg = 87.2 °C) are outside of it. Additionally, the plasticizer leads to an improved solubility in common solvents.
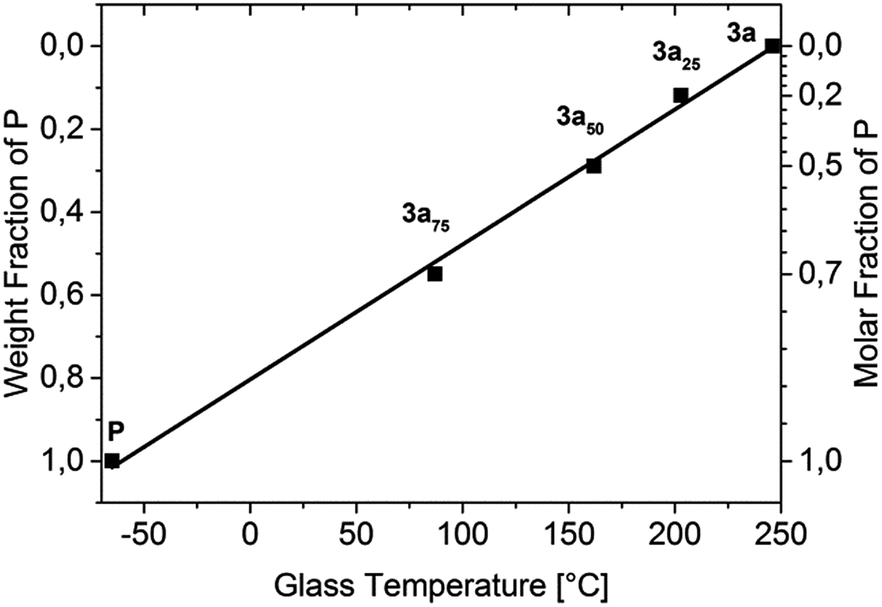 | ||
| Fig. 1 T g dependence on the weight and molar fractions of P in the polymers 3a, 3a25, 3a50, and 3a75. | ||
Note that the introduction of the plasticizer leads to no significant changes of the HOMO and LUMO levels of the polymers (see Table 2). However, the electrical hole current in unipolar diodes (not shown) is considerably impaired for those polymers as compared to the currents in polymer layers without plasticizer. This may be due to the insulating nature of the decylbenzene moieties.
Crosslinking of low- and high-Tg polymers
In order to demonstrate that the glass transition affects the crosslinking kinetics strongly, 1-(azidomethyl)-4-vinylbenzene and monomer 2a were copolymerized either with or without additional plasticizer P to give a crosslinkable low- (3a43AZ15) and high-Tg (3aAZ15) polymer with 15 M% azide content, respectively (Scheme 3). Crosslinking can be performed thermally after adding 4,4′-(propane-2,2-diyl)bis((prop-2-yn-1-yloxy)-benzene) (XL).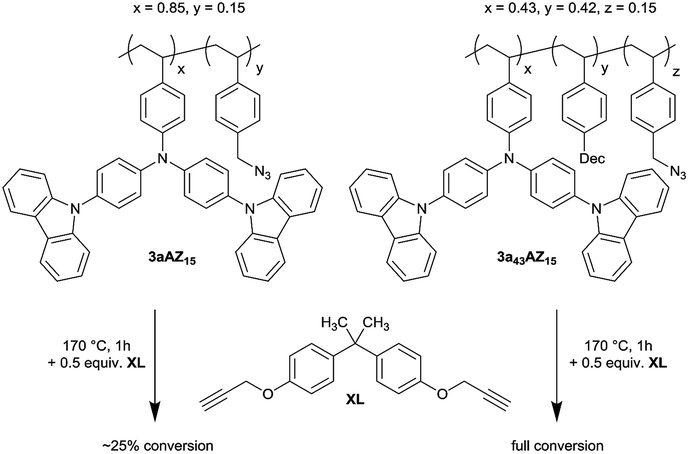 | ||
| Scheme 3 Crosslinker incorporation into low and high Tg polymers. | ||
Crosslinking of copolymerized azides and alkynes in poly-styrenes via triazole formation has initially been described by Spruell et al.14 The introduction of bis-alkyne (XL) as separate crosslinker component has the advantage of an improved bench-stability of the resulting materials. The thermal range of crosslinking can be between 150 °C, where the cycloaddition of azides and alkynes starts, and 200 °C, where unwanted thermal decomposition of the azides and nitrene formation set in. Formed nitrenes can then easily insert into aliphatic and aromatic bonds with detrimental effects on the charge transport in conjugated materials.15
Azide decomposition is visible as an exothermic reaction in differential scanning calorimetry (DSC) spectra of neat 3aAZ15 and 3a43AZ15 at around 250 °C (Fig. S1 and S2†). The glass transitions of both polymers are visible in the subsequent DSC cycle at 250 °C and 172 °C, respectively. The reduced Tg of 3a43AZ15 is clearly due to the incorporation of the plasticizer. Addition of the crosslinker XL to 3aAZ15 and 3a43AZ15 leads to an exothermic DSC peak above 150 °C which can be ascribed to the desired azide-alkyne cycloaddition. After the cycloaddition, the Tgs of the crosslinked polymers are further reduced to 240 °C and 158 °C, respectively. This decrease can be explained by a further plasticizing effect of XL.
Performing the crosslinking reaction at 170 °C in the presence of an equal amount of azide and alkyne moieties leads, however, to vastly different azide conversions for 3aAZ15 and 3a43AZ15. The azide-alkyne reaction can be followed with isothermal ATR-FTIR spectroscopy (Fig. S3 and S4†) by recording the corresponding symmetrical-stretching mode bands (N3, 2095 cm−1; C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H, 3300 cm−1). Fig. 2 shows the azide-related peak areas over time during constant heating at 170 °C. While the crosslinking reaction is complete (i.e., 100% azide consumption) after 35 min for low-Tg3a43AZ15, the azide conversion in 3aAZ15 stops after only a few minutes and ca. 75% of the azide groups remain unreacted. The latter is due to the high glass transition of 3aAZ15 which hinders the segmental movement of the crosslinker groups and therefore prevents the encounter of the reaction partners. It is thus clear that an effective crosslinking procedure must be conducted above the glass transition of the polymer, but well below the azide decomposition temperature. For 3aAZ15 (i.e., the high-Tg polymer without plasticizers) both conditions cannot be fulfilled at the same time, which highlights exemplary the importance of glass transition tuning presented in this work.
C–H, 3300 cm−1). Fig. 2 shows the azide-related peak areas over time during constant heating at 170 °C. While the crosslinking reaction is complete (i.e., 100% azide consumption) after 35 min for low-Tg3a43AZ15, the azide conversion in 3aAZ15 stops after only a few minutes and ca. 75% of the azide groups remain unreacted. The latter is due to the high glass transition of 3aAZ15 which hinders the segmental movement of the crosslinker groups and therefore prevents the encounter of the reaction partners. It is thus clear that an effective crosslinking procedure must be conducted above the glass transition of the polymer, but well below the azide decomposition temperature. For 3aAZ15 (i.e., the high-Tg polymer without plasticizers) both conditions cannot be fulfilled at the same time, which highlights exemplary the importance of glass transition tuning presented in this work.
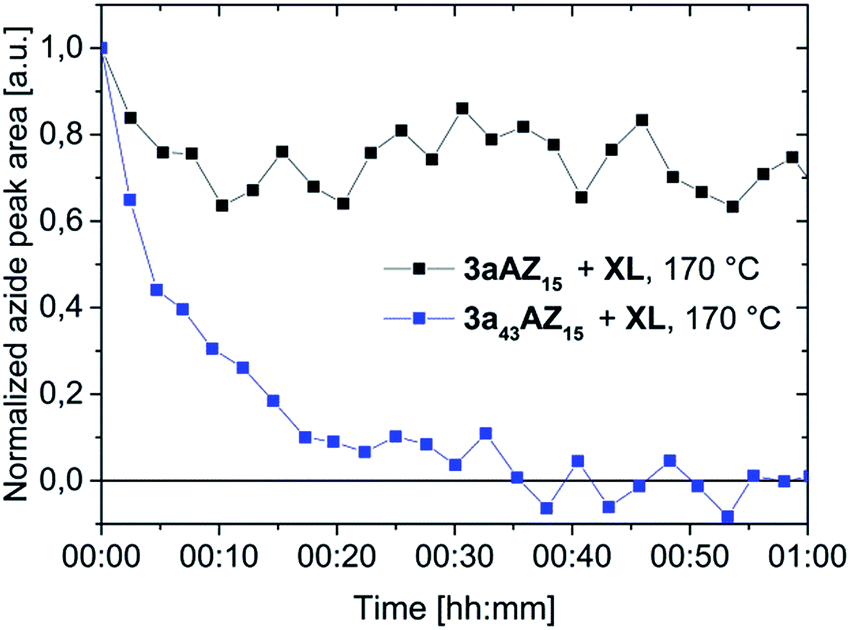 | ||
| Fig. 2 Peak area isotherm of the 2095 cm−1 symmetric azide stretching at 170 °C of polymers 3aAZ15 and 3a43AZ15 in the presence of the crosslinker XL. | ||
Notice that we observe a significant (∼20%) wash-off of the (partially) crosslinked 3aAZ15 layers after rinsing with toluene (see UV-Vis absorption spectra Fig. S5†). Crosslinked 3a43AZ15 layers, on the contrary, exhibit excellent solvent resistance (Fig. S6†).
Simultaneous HOMO level and Tg tuning by alkoxy groups with varied alkyl lengths
An alternative way to achieve a Tg adaption is the direct substitution of the pendant carbazole groups (Scheme 4). We decided to introduce alkoxy groups at different positions on the carbazole groups of the poly-TCTA (3a) to reduce the Tg while additionally increasing the electron density at the carbazole. The latter approach gives a viable ability to fine-tune the HOMO level of the polymer, as has been shown previously in a study on substituted molecular TAPC.12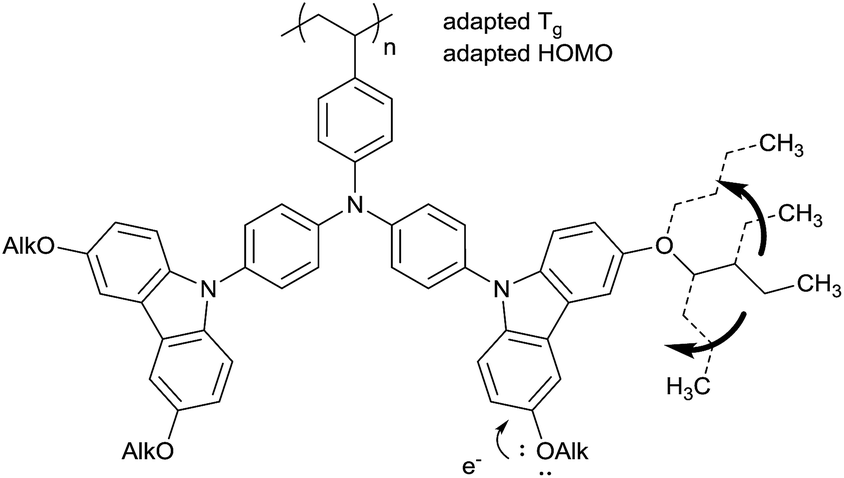 | ||
| Scheme 4 The plasticizing and electronic influence of alkoxy groups on poly-TCTA. | ||
For the alkoxy-substituted TCTA polymers (3b–f) an increasing number of alkylic carbons per pendant group were introduced. The alkoxy substitution of carbazoles (b/d/e/f, Scheme 5) was performed following a procedure by M. A. Keegstra et al. for the copper(I)halide catalyzed synthesis of alkyl aryl ethers16 which was applied to carbazoles before.17,18 2-((2-Ethylhexyl)oxy)-9H-carbazole (c) was obtained by simple etherification of the aryl ether and the corresponding 2-hydroxy-9H-carbazole. The following reactions towards the polymers 3b–f (top of Scheme 5) were performed according to a procedure published by Thesen et al.8 The incorporation of the alkoxy groups leads to a notable decrease of Tg (see Table 1). As compared to unsubstituted poly-TCTA, there is a minor Tg reduction of 26 K for polymer 3b with two methoxy groups, a 62 K decrease for polymer 3e with four ethoxy groups, and a decrease of 100 K for polymers 3c, 3d, and 3f with four butoxy or two ethylhexyloxy substituents. As a rule of thumb, each additional alkylic carbon per pendant group reduces Tg by about 7–13 K. Polymers 3c, 3d, and 3f have glass transitions at around 150 °C which we prefer for the use in OLEDs as outlined earlier.
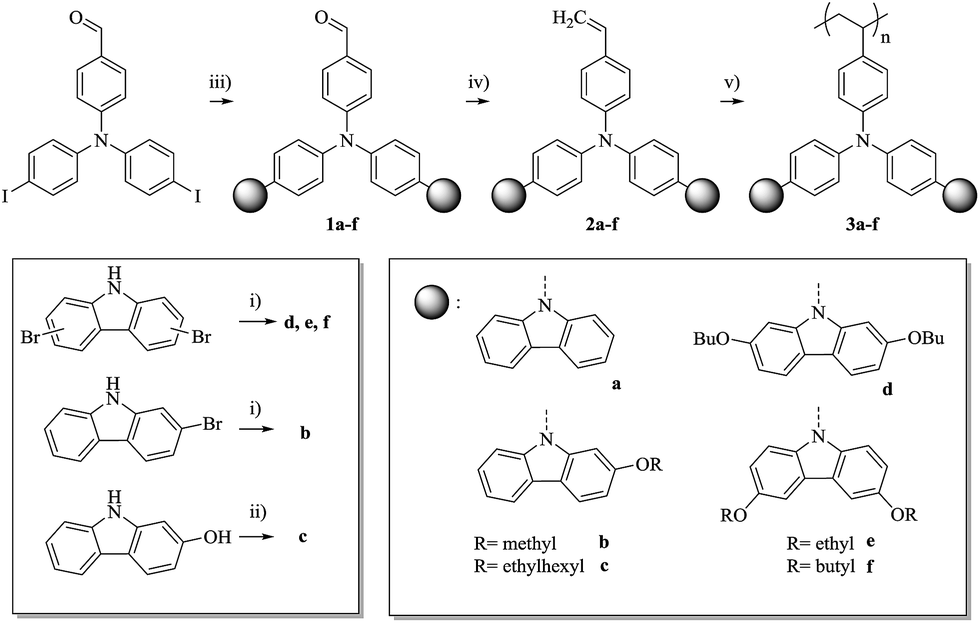 | ||
| Scheme 5 Synthetic route towards Tg-adapted poly-TCTA copolymers and alkoxy-substituted poly-TCTA derivatives with improved hole injection properties. (i) NaOAlk, bromo-9H-carbazole, CuI, DMF, 120 °C, 1–24 h; (ii) 9H-carbazol-2-ol, 3-(bromomethyl)heptane, K2CO3, DMF, 100 °C, 20 h; (iii) a–f, Cu-bronze, K2CO3, 18-crown-6, o-DCB, 200 °C, 1 h to 24 h; (iv) 1a–f, MePPh3Br, KOtBu, 0 °C, 2 h; (v), AIBN (2 M%), THF (toluene for 3f), 50 °C, 2a–f. | ||
Polymers 3a, 3a25, 3a50, and 3a75 have degrees of polymerization (DP) of around 50–70 and polymers 3b–f of around 15–40 (Table 1). From our experience, the Tg values of polystyrenes substituted with bulky side groups are essentially independent of molecular weight, if DP ≥ 15. This is supported by Claudy et al.19 who show that the Tg of polystyrene remains almost unchanged for polymer chains with more than 25 repeat units. The azide functionalized polymers 3aAZ15, 3a43AZ15 and 3fAZ15 have similar weight distributions compared to the non-crosslinkable counterparts (Table 1).
In addition to the adaption of the glass transition, the introduction of alkoxy groups at the functional π conjugated carbazoles allows for a tuning of the energetic states. Here, we investigate this concept by introducing substituents with varied alkyl chain lengths and varied substitution positions. HOMO level values were determined with photoelectron spectroscopy in air (PESA) and cyclovoltammetry (CV); these results are in good agreement (with a maximum deviation of 0.15 meV for polymer 3a25) and are summarized in Table 2. All polymers were reversible for several CV-measurement cycles, indicating a good electrochemical stability. The unsubstituted polymer 3a has the deepest HOMO level, followed by the polymers bearing alkoxy groups in the meta-position of the carbazole nitrogen (3b/3c/3d). Unexpectedly, the work function change for 3b with one methoxy group (Δ = 0.13 eV, relative to 3a and according to PESA measurements) is slightly bigger than that for 3c (having one ethoxyhexyl group, Δ = 0.04 eV) and 3d (having two butoxy groups, Δ = 0.09 eV). Generally, however, the influence of meta-substituted alkoxy groups on the HOMO level is relatively small and the differences are close to the uncertainty of the PESA and CV measurements. A significant change of Δ = 0.3 eV is observed for the HOMO levels of polymers 3e and 3f which have their alkoxy substituents in para-position to the carbazole-nitrogen. This can be explained by para-ring substituent effects.
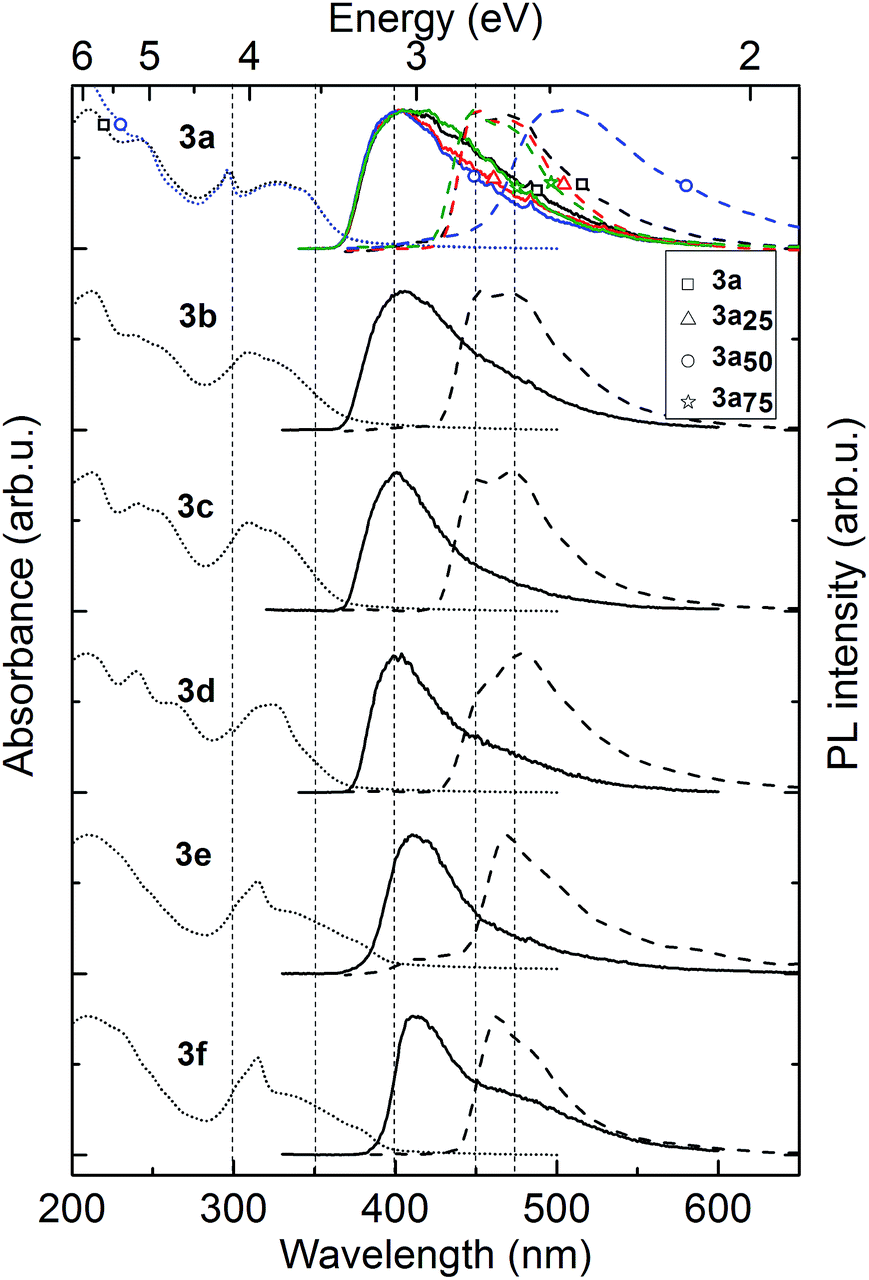 | ||
| Fig. 3 UV/Vis (dotted lines) and photoluminescence (PL) spectra (straight lines) and phosphorescence (dashed lines) of the polymers 3a–f. The top-graph shows additionally the spectra of 3a25 (triangle), 3a50 (circle) and 3a75 (star) for comparison. | ||
The phosphorescence spectra of each material act in a similar fashion to fluorescence. 3a–d demonstrate small changes in phosphorescence spectra, the spectra of polymers 3e and 3f are redshifted by 10 nm. Polymer 3e is the only material showing a small delayed fluorescence signal which might indicate enhanced triplet mobility or higher ISC yield. Most importantly, all polymers have a high triplet energy (2.8–2.9 eV, see Table 2) which is compatible with their use as host or hole-transporting material in OLEDs with green-to-blue phosphorescent emitters.
The various substituents on the polymers have very little effect on the triplet energy, as desired. However, polymer 3a50 containing 50 M% of 1-decyl-4-vinylbenzene spacer demonstrated very broad, structureless and redshifted, by 50 nm, phosphorescence. This we ascribe to emission from triplet states dimers, formed between interacting neighbouring carbazole units, as was previously observed in PVK.22
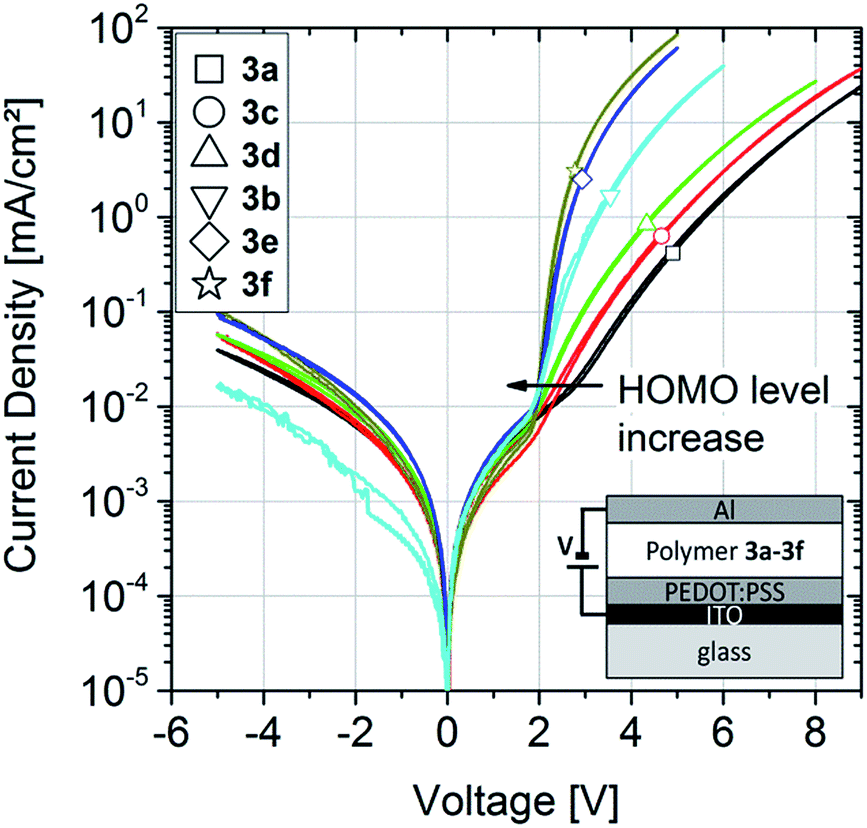 | ||
| Fig. 4 Electrical characteristics of hole-only diodes prepared from polymers 3a–f. Positive voltages refer to the injection of holes from the ITO contact into the polymer layer. The polymer layers have similar thicknesses of 80–100 nm. Inset: sample structure of the hole-only diodes. | ||
In order to represent the full span of the HOMO level range which is accessible with the new TCTA-based polymers, we tested the crosslinkable versions of 3a (unsubstituted poly-TCTA with 15 M% azide content, 3a43AZ15) and 3f (poly-3,6-(BuO)4TCTA with 15 M% azide content, 3fAZ15) as hole-transport layers in fully solution-processed OLEDs (i.e., solution-processing except for the electrodes). Notice that in the case of unsubstituted poly-TCTA, we have used the low-Tg variant with decyl styrene plasticizer in order to allow for crosslinking well below 200 °C.
15–20 nm thick hole-transport layers were fabricated by spin-coating from toluene solutions on top of ITO/PEDOT:PSS patterned electrodes. Crosslinking of these layers was performed by heating at 170 °C for 40 min. The emission layer is a ternary blend consisting of an iridium-based phosphorescent green emitter, a co-host, and an electron transporting polymer as is described in our previous work, ref. 23 and 24 The emission layer was annealed at 180 °C for 30 min. The Ba/Ag cathode was deposited by subsequent evaporation. Further details of the sample preparation can be found in the ESI.†
The OLED characteristics are shown in Fig. 5a. Clear diode behaviors and high rectification ratios are observed. Comparing the devices with 3fAZ15 and with 3a43AZ15 HTL, the current and luminance onset voltage is increased for the latter sample due to the deep HOMO level of 3a43AZ15, as expected.
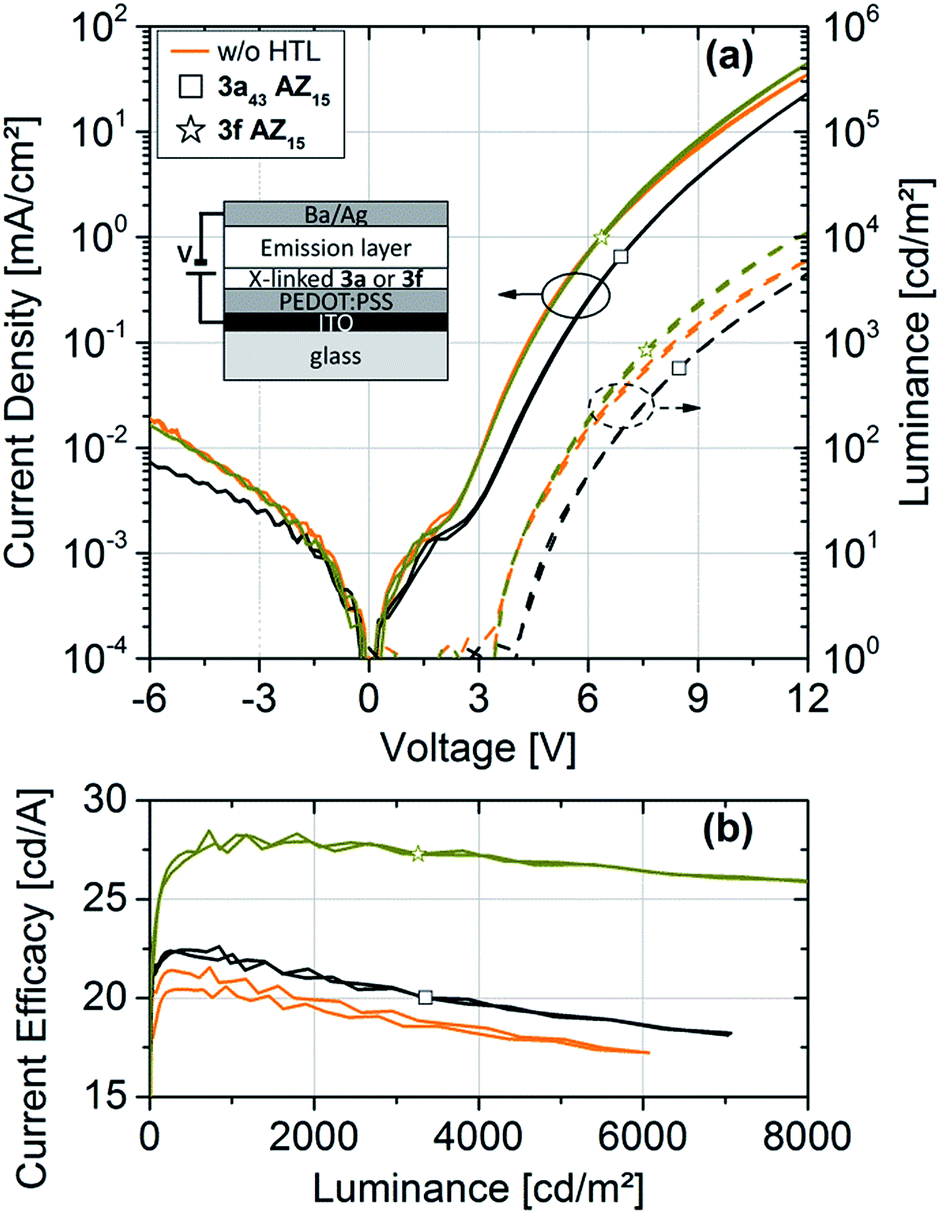 | ||
| Fig. 5 (a) j–L–V characteristics and (b) current efficacies of OLEDs with and without hole-transport layers based on the crosslinkable polymers 3a or 3c with incorporated azide (AZ) moieties. | ||
Fig. 5b shows the current efficacies of the devices with and without HTL. It can be clearly seen that the use of a HTL, separating the emission layer from the anode, leads to an efficacy increase as compared to devices with no HTL. The highest current efficacy is exhibited by the device with 3fAZ15 HTL, which is probably due to the proper adaption of its HOMO level to the energetic landscape of the emissive layer.24 We cannot determine the detailed physical role that the HTL plays in our devices within the scope of this article; however, it seems plausible that the high triplet energies and the high-lying LUMO-derived states of the TCTA-based polymers serve for a blocking of triplets and electrons, such that those species become confined to the emission layer. Most importantly, we have shown that both crosslinkable TCTA-based polymers are suitable and advantageous to be used as HTL materials in OLEDs as they enable the subsequent solution-processing of the emission layer without any re-dissolution.
Conclusions
We have demonstrated two approaches to adapt functional side-chain polystyrenes towards the needs of a solution-processed multilayer OLED structure: by introduction of plasticizers as co-monomers or as substituents to the π-conjugated units of the prototypical hole-transporting TCTA side-chain polystyrene, the glass transition can be tuned in a wide range between 87 and 245 °C. This is of particular importance for thermal crosslinking reactions, where the reaction temperature should be far lower than 200 °C in order to enable a cost-efficient, complete process and to avoid decomposition of the reactants. With the moderate glass transitions of the new poly-TCTAs at around 150 °C high conversions can be achieved upon thermally initiated crosslinking, which is demonstrated on crosslinkable polymers containing an azide-alkyne crosslinker.Moreover, the approach of substituting the TCTA core with alkoxy groups allows for a concomitant tuning of the HOMO level, which is desired for the application of these polymers as hole transport layers in OLED structures. While the HOMO level can be adjusted to values between −5.29 and −5.62 eV, the high LUMO level position and the high triplet energy of poly-TCTA are maintained. These properties render the crosslinkable, substituted poly-TCTA variants ideal to be used as hole-transport electron-blocking layer between the anode and the emission zone of green phosphorescent OLEDs, as we have demonstrated for solution-processed devices.
Acknowledgements
F. R. P. Limberg, P. Pingel, S. Janietz, and H. Krüger acknowledge funding by the German BMBF (ENAB-SPOLED 13N23634).References
- B. Zhang, L. Liu and Z. Xie, Isr. J. Chem., 2014, 54, 897–917 CrossRef CAS.
- W. Li, J. Li and M. Wang, Isr. J. Chem., 2014, 54, 867–884 CrossRef CAS.
- S. Sax, N. Rugen-Penkalla, A. Neuhold, S. Schuh, E. Zojer, E. J. W. List and K. Müllen, Adv. Mater., 2010, 22, 2087–2091 CrossRef CAS PubMed.
- C. W. Joo, S. O. Jeon, K. S. Yook and J. Y. Lee, Org. Electron., 2009, 10, 372–375 CrossRef CAS.
- C. A. Zuniga, S. Barlow and S. R. Marder, Chem. Mater., 2011, 23, 658–681 CrossRef CAS.
- G. Liaptsis, D. Hertel and K. Meerholz, Angew. Chem., 2013, 125, 9742–9746 CrossRef.
- M.-H. Wu, J.-H. Lee, M.-K. Leung, C.-C. Liao and Y. Chang, Proc. SPIE 5519, Organic Light-Emitting Materials and Devices VIII, ed. Z. H. Kafafi and P. A. Lane, 2004, vol. 263, DOI:10.1117/12.559561.
- M. W. Thesen, B. Höfer, M. Debeaux, S. Janietz, A. Wedel, A. Köhler, H.-H. Johannes and H. Krueger, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3417–3430 CrossRef CAS.
- X. Gong, S.-H. Lim, J. C. Ostrowski, D. Moses, C. J. Bardeen and G. C. Bazan, J. Appl. Phys., 2004, 95, 948–953 CrossRef CAS.
- S. Tokito, M. Suzuki, F. Sato, M. Kamachi and K. Shirane, Org. Electron., 2003, 4, 105–111 CrossRef CAS.
- G. Greczynski, T. Kugler, M. Keil, W. Osikowicz, M. Fahlman and W. R. Salaneck, J. Electron Spectrosc. Relat. Phenom., 2001, 121, 1–17 CrossRef CAS.
- G. Liaptsis and K. Meerholz, Adv. Funct. Mater., 2013, 23, 359–365 CrossRef CAS.
- C. G. Overberger, C. Frazier, J. Mandelman and H. F. Smith, J. Am. Chem. Soc., 1953, 75, 3326–3330 CrossRef CAS.
- J. M. Spruell, M. Wolffs, F. A. Leibfarth, B. C. Stahl, J. Heo, L. A. Connal, J. Hu and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 16698–16706 CrossRef CAS PubMed.
- R.-Q. Png, P.-J. Chia, J.-C. Tang, B. Liu, S. Sivaramakrishnan, M. Zhou, S.-H. Khong, H. S. O. Chan, J. H. Burroughes, L.-L. Chua, R. H. Friend and P. K. H. Ho, Nat. Mater., 2010, 9, 152–158 CrossRef CAS PubMed.
- M. A. Keegstra, T. H. A. Peters and L. Brandsma, Tetrahedron, 1992, 48, 3633–3652 CrossRef CAS.
- Y. Kikugawa, Y. Aoki and T. Sakamoto, J. Org. Chem., 2001, 66, 8612–8615 CrossRef CAS PubMed.
- B. Schmidt, S. Krehl, A. Kelling and U. Schilde, J. Org. Chem., 2012, 77, 2360–2367 CrossRef CAS PubMed.
- P. Claudy, J. M. Létoffé, Y. Camberlain and J. P. Pascault, Polym. Bull., 1983, 9, 208–215 CrossRef CAS.
- D. Gudeika, J. V. Grazulevicius, G. Sini, A. Bucinskas, V. Jankauskas, A. Miasojedovas and S. Jursenas, Dyes Pigm., 2014, 58–70, DOI:10.1016/j.dyepig.2014.02.023.
- A. Tomkeviciene, J. V. Grazulevicius, K. Kazlauskas, A. Gruodis, S. Jursenas, T. H. Ke and C. C. Wu, J. Phys. Chem. C, 2011, 4887–4897, DOI:10.1021/jp111333v.
- V. Jankus and A. P. Monkman, Adv. Funct. Mater., 2011, 21, 3350–3356 CrossRef CAS.
- B. C. D. Salert, A. Wedel, L. Grubert, T. Eberle, R. Anémian and H. Krueger, Adv. Mater. Sci. Eng., 2012, 2012, 1–15 CrossRef.
- B. C. D. Salert, H. Krueger, S. A. Bagnich, T. Unger, F. Jaiser, M. Al-Sa'di, D. Neher, A. Hayer and T. Eberle, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 601–613 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12963j |
| This journal is © The Royal Society of Chemistry 2015 |