
Insight into the origin of ferromagnetism in Fe-doped ZnO diluted magnetic semiconductor nanocrystals: an EXFAS study of local structure†
Abstract
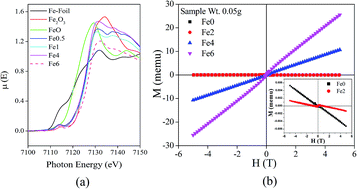
In this paper we have studied the structural, local structural and magnetic properties of sol–gel derived Zn1−xFexO (0 ≤ x ≤ 0.06) nanoparticles. The crystalline structure and crystallite size have been estimated by X-ray diffraction with Rietveld refinement and high-resolution transmission electron microscopy (HRTEM). Other structural and local structural properties have been studied by extended X-ray absorption fine structure (EXAFS)-, X-ray absorption near edge structure (XANES)- and Raman-analysis. Weak ferromagnetism is observed at room temperature and magnetization increases with increasing Fe-concentration. The oxygen vacancy assisted bound magnetic polarons (BMPs) and possibly the grain boundaries are responsible for this room temperature ferromagnetism. Variation of resistivity with temperature has also been studied. Appearance of ferromagnetism in ZnO : Fe nanoparticles may open the potential in bio-imaging and drug-delivery applications.
 Please wait while we load your content...
Please wait while we load your content...

