
The origin for highly enantioselective induction of 1-naphthol to isatin-derived N-Boc ketimines catalyzed by quinine thiourea catalyst: an experimental and computational study†
Prathibha kumariab,
Sunirmal Barikbc,
Noorul H. Khan*ab,
Bishwajit Ganguly*bc,
Rukhsana I. Kureshyab,
Sayed H. R. Abdiab and
H. C. Bajajab
aDivision of Inorganic Materials and Catalysis, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), Council of Scientific & Industrial Research (CSIR), G. B. Marg, Bhavnagar, 364002, Gujarat, India. E-mail: khan251293@yahoo.in; Fax: +91-278-2566970; Tel: +91-278-2567760
bAcademy of Scientific and Innovative Research (AcSIR), (CSIR-CSMCRI), Bhavnagar, 364002, Gujarat, India
cAnalytical Discipline and Centralised Instrumental Facility, CSIR, Central Salt and Marine Chemicals Research Institute (CSMCRI), G. B. Marg, Bhavnagar 364 021, Gujarat, India. E-mail: ganguly@csmcri.org
First published on 10th August 2015
Abstract
An enantioselective aza-Friedel–Crafts reaction of 1-naphthol with isatin derived N-Boc ketimines by cinchona based bifunctional thiourea as organo-catalyst is reported. In general the derivatives of Betti base are formed in excellent enantioselectivities (95–99%) with high yields (<99%). The combination of experimental and computational studies have revealed the origin of the stereoselectivity of the aza-Friedel–Crafts reaction of 1-naphthol with isatin derived N-Boc ketimines by cinchona based bifunctional thiourea as organo-catalyst. The attractive electrostatic interactions in the Re face transition state and the deleterious lone-pair⋯π interactions in the Si face transition state governed the formation of Re-face as a major product in this aza Friedel–Crafts reaction. The NMR studies performed to examine the formation of the complex with 1-naphthol, isatin derived N-Boc ketimines and cinchona based bifunctional thiourea catalyst has corroborated the calculated complex geometry employed in the study.
Introduction
The Friedel–Crafts reaction has undoubtedly been positioned as one of the most important carbon–carbon bond forming reactions for the synthesis of valuable building blocks of biologically active molecule.1 At the beginning of the 20th century, Mario Betti discovered the three-component reaction of 2-naphthol, aryl aldehydes and ammonia or amines for the synthesis of aminobenzylnaphthols,2 known as the Betti reaction and the product as a Betti base.3 Aminonaphthols have been used in the synthesis of antibacterial, hypotensive, and brady-cardiac activities4–6 and also as ligands in metal catalysed asymmetric reactions.7,8 Further, biologically active natural products and pharmaceuticals9 like 3-amino-2-oxindoles can be obtained by aza Henry,13 Strecker,14 allylation and Friedel–Crafts reactions10–12,15 of isatin derived ketimines. For the present study, we used cinchona based bifunctional thiourea as organocatalyst for Friedel–Crafts alkylation of isatin N-Boc ketimine with 1-naphthol to form Betti base in excellent yield and ee. We have also explored different N1-substituted isatin N-Boc ketimine and observed products with excellent enantiomeric excess (ee) at lower temperature. The origin of stereoselectivity rendered by the bifunctional thiourea as organocatalyst for Friedel–Crafts alkylation of isatin N-Boc ketimine with 1-naphthol has been revealed by DFT calculations. It is important to note that while we were drafting the present manuscript a paper on similar lines (albeit without mechanistic studies) appeared in the literature.16Results and discussions
Initially, the catalytic ability of cinchona alkaloids 1a (QN), and 1b (QD) was investigated for the Friedel–Crafts reaction of 1-naphthol (6a) with N1-benzylisatin derived N-Boc ketimine (7a) as model substrate in DCM and 4 Å molecular sieves at room temperature. The desired product 8a was isolated in good yield, but with poor enantioselectivity (Table 1, entries 1 and 2). The same reaction when performed with different chiral di-amine and amino alcohol based thiourea organocatalysts (2a, 2b, 3a, 4a), the desired product 8a was obtained with low enantioselectivity (Table 1, entries 3–6). In order to improve the activity and enantioinduction in the product 8a we next, investigated the catalytic activity of 9-thiourea derivatives of cinchona alkaloids (Table 1, entry 7 and 8), where the quinine thiourea 5b provided the Friedel–Crafts adduct 8a in excellent yield (98%) and enantiomeric excess (99%) (Table 1, entry 8).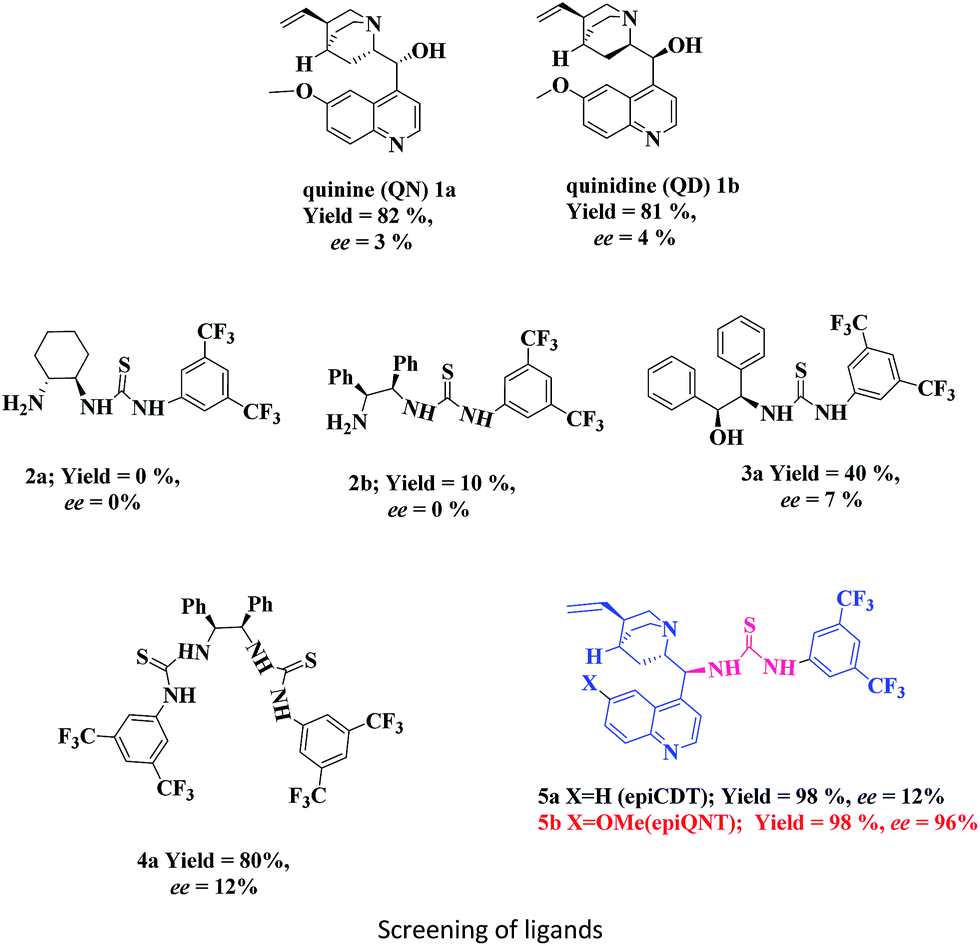
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | eec (%) |
| a All the reactions were carried out by using 1-naphthol (3 mmol), N1-benzylisatin ketimine (0.2 mmol), and catalyst (5 mol%) in DCM at 25 °C.b Isolated yields after column chromatography.c ee determined by chiral HPLC using Daicel Chiralpak IA column. | ||||
| 1 | 1a | 8 | 82 | 3 |
| 2 | 1b | 8 | 81 | 4 |
| 3 | 2a | 8 | 0 | 0 |
| 4 | 2b | 8 | 10 | 0 |
| 5 | 3a | 8 | 40 | 7 |
| 6 | 4a | 8 | 80 | 12 |
| 7 | 5a | 8 | 98 | 12 |
| 8 | 5b | 8 | 99 | 99 |

In order to promote the optimization of reaction condition we next moved to see the effect of different solvent for the reaction of 1-naphthol with ketimine 7a. Variation of solvents showed the disparity in enantioselectivity of the desired product 8a (Table 2). The non-polar solvents such as toluene and xylene were ended the reaction with good yield as well as enantioselectivity (Table 2, entries 1, 2). However with polar aprotic solvents such as ethyl acetate provided the product 8a in good yield but moderate enantioselectivity (Table 2, entry 3). Among the ethereal solvents like THF, dioxane and diethyl ether provided the desired product 8a in good yields with good to moderate enantioselectivity (Table 2, entries 4–6). Among different chlorinated solvents like chloroform and dichloromethane (DCM) proved as best solvents because it provided the product 8a in good yield and the best enantioselectivity (Table 2, entries 7 and 8). Thus, 5 mol% of catalyst 5b with 4 Å molecular sieves and dichloromethane as solvent at 25 °C was found to be optimum providing the Friedel–Crafts adduct 8a with 99% yield with 99% ee (Table 2, entry 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Time (h) | Yieldb (%) | eec (%) |
| a All the reactions were carried out by using 1-naphthol (3 mmol), benzylisaitn ketimine (0.2 mmol), and catalyst (5 mol%) in different solvents mentioned in table at 25 °C.b Isolated yields after column chromatography.c ee determined by chiral HPLC using Daicel Chiralpak IA column. | |||||
| 1 | Toluene | 25 | 6 | 95 | 94 |
| 2 | Xylene | 25 | 8 | 80 | 83 |
| 3 | Ethylacetate | 25 | 14 | 90 | 92 |
| 4 | THF | 25 | 8 | 92 | 86 |
| 5 | Dioxane | 25 | 8 | 55 | 65 |
| 6 | Diethylether | 25 | 8 | 70 | 50 |
| 7 | Chloroform | 25 | 5 | 95 | 80 |
| 8 | DCM | 25 | 4 | 99 | 99 |
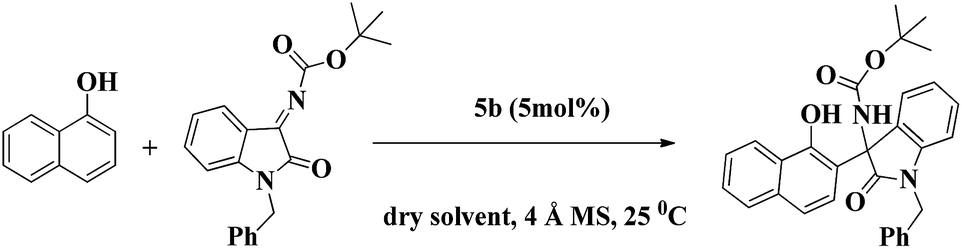
With the optimum reaction conditions (Table 2, entry 8), the scope of the organocatalyst 5b was further extended for the asymmetric F–C reaction of various substituted isatin ketimines at N1 position of isatin and substitutions at benzene ring of isatin with 1-naphthol (Table 3). The results have shown that the present catalytic system is highly efficient in term of excellent yield (99%) and ee (95–99%) irrespective of electron donating (5-methoxy) or withdrawing (5-nitro, 5-fluoro, 7-fluoro, 5-chloro, 5-bromo, 6-bromo) groups and the position of the substituents on isatin ketimines. On conducting the same reaction with substituted 1-naphthol having electron donating (4-methoxy) and electron withdrawing (4-chloro) their corresponding products 8m and 8n were achieved with excellent enantioselectivies and yields. However, on performing the reaction of N1-benzylisatin derived N-Boc ketimine with 2-naphthol, the corresponding product 8n was achieved in excellent yield but with moderate ee (yield 99%, ee 92%) with 10 mol% of catalyst loading.

In order to develop a useful understanding and to ascertain the precise role of catalyst 5b asymmetric Friedel–Crafts reaction of N1-methylisatin derived N-Boc ketimine as representative substrate, we have conducted a series of 1H and 13C NMR experiments in CDCl3, for looking the interaction of catalyst 5b with the substrate 7b. When we mixed the catalyst 5b with N1-methylisatin derived N-Boc ketimine 7b in presence of 1-naphthol 6 (Fig. 1 and 2) we have observed that out of the two protons (Ha, Hb) appeared at δ 5.68 and δ 9.94 in the catalyst, the Hb proton after interaction with carbonyl oxygen of isatin ketimine showed more down field shift and lie at δ 10.05 as compared to Ha (weakly coordinated) to ester and very little down field shift (0.002 ppm) was observed as shown in the Fig. 1. Besides, there was also a down field shift seen in the proton H9 present adjacent to the Ha proton. This indicates that the substrate ketimine is more closely interacted with the two N–H protons Ha and Hb of the catalyst 5b and more likely to form an intermediate I in the proposed catalytic cycle (Scheme 2). Further, an up field shift in Ha and Hb protons of the catalyst was observed, when 1-naphthol is mixed with the fresh catalyst 5b which may be due to the electron density provided by oxygen of the 1-naphthol as shown in the Scheme 1 and correspond to the intermediate II (Scheme 2).
 | ||
| Fig. 1 All the reactions were carried out in CDCl3 and recorded the 1H NMR. [A] 1H NMR of fresh catalyst, [B] 1H NMR of the catalyst after interaction with ketimine for 15 min, [C] 1H NMR of the catalyst after interaction with 1-naphthol for 15 min. | ||
 | ||
| Fig. 2 All the reactions were carried out in CDCl3 and recorded the 13C NMR. [A] 13C NMR of fresh catalyst, [B] 13C NMR of the pure N1-methylisatin derived N-Boc ketimine, [C] 13C NMR of the catalyst after interaction with N1-methylisatin derived N-Boc ketimine for 15 min. [D] 13C NMR of the catalyst after interaction with 1-naphthol for 15 min. | ||
 | ||
| Scheme 1 Activation sites of the bi-functional catalyst with the substrates. | ||
 | ||
| Scheme 2 Proposed catalytic cycle. | ||
These observations are inconsonance with the earlier report.17 A similar trend was also observed in 13C NMR of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –CN of ketimine before and after interaction with the catalyst 5b via hydrogen bonding. Upon addition of 1-naphthol to the catalyst there was a drastic shift at C6, C8 and C9 positions of the catalyst (Fig. 2). Hence we predicted that the –OH of the 1-naphthol was activated via hydrogen bonding with the tertiary nitrogen present in the thiourea. Based on these experimental data we have proposed the catalytic cycle of this reaction as shown in the Scheme 2.
O and –CN of ketimine before and after interaction with the catalyst 5b via hydrogen bonding. Upon addition of 1-naphthol to the catalyst there was a drastic shift at C6, C8 and C9 positions of the catalyst (Fig. 2). Hence we predicted that the –OH of the 1-naphthol was activated via hydrogen bonding with the tertiary nitrogen present in the thiourea. Based on these experimental data we have proposed the catalytic cycle of this reaction as shown in the Scheme 2.
Kinetic studies
To understand the mechanism of the F–C alkylation of isatin ketimine, kinetic experiments were performed with N1-benzylisatin derived N-Boc ketimine 7a as a model substrate and as a function of the concentration of catalyst 5b and substrate. In all the kinetic runs, the plots of formation of the F–C alkylated product with time was found to be linear in the beginning of the reaction and attained saturation near its completion (Fig. 3). On the basis of this observation, the initial rate constants, kobs were determined by directly estimating the amount of alkylated product formed up to the completion of the reaction.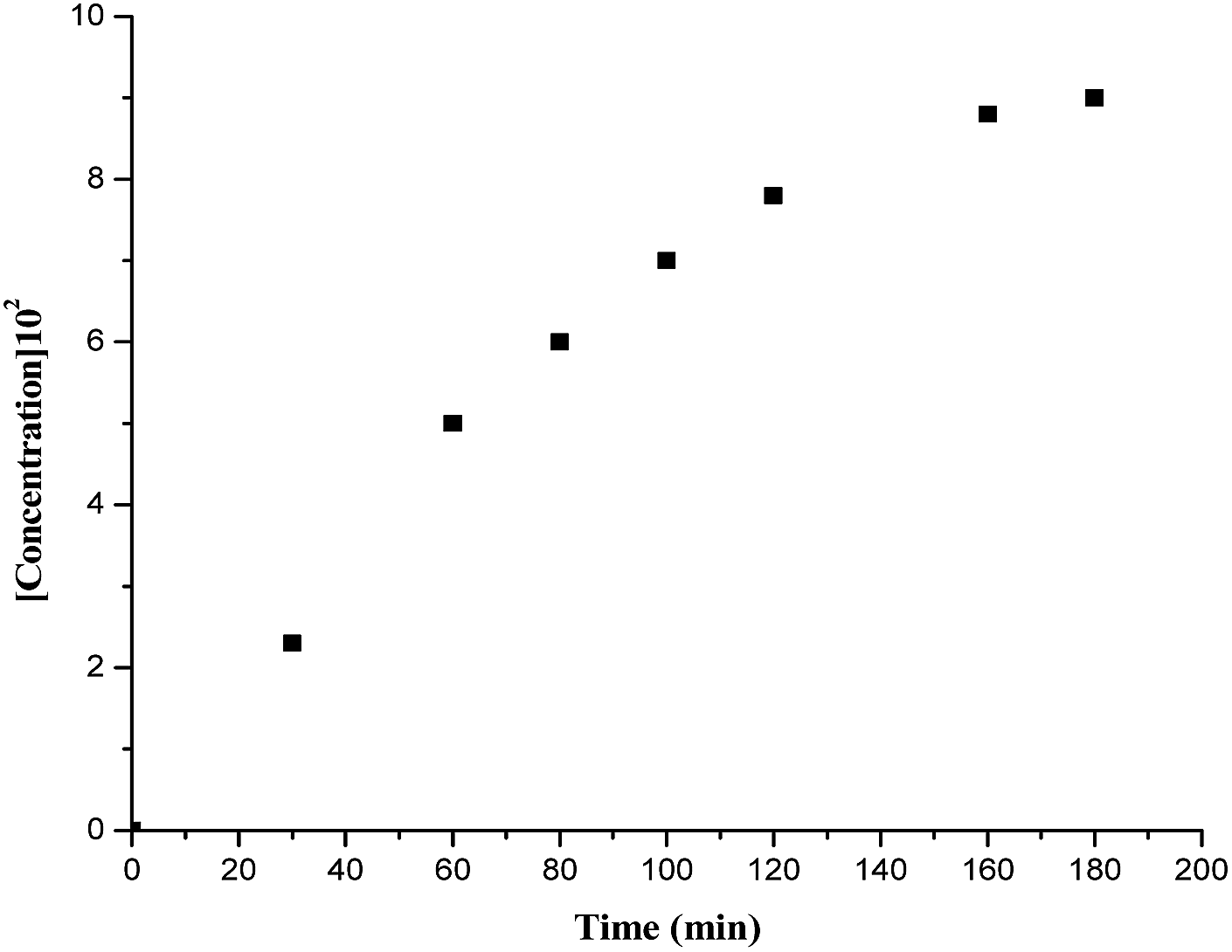 | ||
| Fig. 3 Plot of time versus concentration of product at 25 °C, [catalyst 5b] = 0.00375 M, [ketimine] = 0.15 M. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kobs/dlog[catalyst] ∼ 1) was obtained, which passes through the origin, indicating that the F–C reaction is of first-order with respect to the concentration of the catalyst 5b (Fig. 4).
kobs/dlog[catalyst] ∼ 1) was obtained, which passes through the origin, indicating that the F–C reaction is of first-order with respect to the concentration of the catalyst 5b (Fig. 4).
| [Cat] × 103 M | [Ketimine] × 102 M | [1-Naphthol] × 102 | kobs × 105 mM min−1 | |
|---|---|---|---|---|
| Catalyst variation | 3.75 | 15 | 22.5 | 4.89 |
| 6.25 | 15 | 22.5 | 7.16 | |
| 7.50 | 15 | 22.5 | 10 | |
| Ketimine variation | 6.25 | 15 | 22.5 | 3.8 |
| 6.25 | 20 | 22.5 | 5.48 | |
| 6.25 | 25 | 22.5 | 7.76 | |
| 6.25 | 30 | 22.5 | 9.98 | |
| Naphthol variation | 6.25 | 15 | 22.5 | 10.47 |
| 6.25 | 15 | 30 | 9.77 | |
| 6.25 | 15 | 37.5 | 10.47 | |
| 6.25 | 15 | 45 | 10.61 |
 | ||
| Fig. 4 Plot of catalyst 5b concentration versus kobs at 25 °C, [ketimine] = 0.15 M, [1-naphthol] = 0.225 M. | ||
kobs/dlog[1-naphthol] ∼ 1) further confirmed the first-order rate dependence on the concentration of 1-naphthol.
 | ||
| Fig. 5 Plot of logkobs verses log[1-naphthol] at 25 °C, [catalyst] = 0.375 × 10−3 M, [ketimine] = 0.15 × 10−2 M. | ||
 | ||
| Fig. 6 Plot of substrate concentration versus kobs at 25 °C, [1-naphthol] = 0.22 × 10−2 M, [catalyst] = 0.375 × 10−3 M. | ||
Computational studies
The reaction mechanism of stereoselective organocatalysis reaction has received a valuable importance in the synthesis of chiral organic compound in diverse field.18 The bifunctional organocatalyst, thiourea tertiary amine organocatalyst has been developed for the simultaneous activation of nucleophile as well as electrophile in the asymmetric catalysis. Such bifunctional chiral organocatalyst attain such a conformational chiral scaffold where the substrate molecules can be activated through relatively weak hydrogen bonding interactions.17 The computational studies help to develop better insight to rationalize the origin of stereoselectivity generated with chiral catalysis and to design more efficient catalytic systems.19 The dual activation of substrate molecules through a bifunctional thiourea-tertiary amine organo catalyst have been well reported.20 To examine the asymmetric aza Friedel–Craft reactions, quinine alkaloid thiourea organo catalyst (Cat.) as a bifunctional organocatalyst has been considered in the DFT calculations. The quinine alkaloid thiourea organo catalyst (Cat.) was used with 1-naphthol as a nucleophile (Nu) and isatin derived N-protected ketimines as a electrophile (EI) in this asymmetric addition reaction.The present study for aza-Friedel–Craft reaction consists of the hybrid thiourea-quinine alkaloid along with the Nu and EI. These molecules are large, hence similar reactions have been examined with hybrid QM/QM21 methods and ONIOM2 methods22 to understand the reaction mechanism and stereoselectivity observed in such cases. We have employed the full density functional method, B3LYP to examine the origin of the stereoselectivity with (Cat.), 1-naphthol as a nucleophile (Nu) and N1-protected isatin derived N-Boc ketimines (EI). The ground state and transition state geometries for all the asymmetric Friedel–Crafts reactions were fully optimized by using B3LYP/6-31G(d) level of theory.23 The single-point energy calculations were performed at M06-2X24 with much higher basis set 6-311+G(d,p)22e by using the ground and transition state structures optimized at B3LYP/6-31G(d) level of theory. The solvent effect was modeled with SCRF-SMD continuum model using dichloromethane.25 The optimized geometries at B3LYP/6-31G(d) level have been used for the frequency calculations to justify that the transition state structures possessing one, and only one, imaginary frequency and no imaginary frequency characterize the ground state geometries. The calculated Gibb's free energies have been reported using B3LYP/6-31G(d) level of theory. All the calculations were performed with the Gaussian 09 suite of program.26
To perform the DFT calculations, first we have optimized the geometry of organocatalyst, quinine thiourea (Fig. 7). The optimized geometry of organocatalyst suggests that the two active sites of chiral catalyst orient in a fashion, where the tertiary nitrogen atom of quinine alkaloid is hydrogen bonded (N–H⋯N) with one of the proton of thiourea moiety (N-Ha, Fig. 7). Such conformation is ideal for the dual activation of both electrophiles and nucleophiles and similar results are reported in the literature.27 This arrangement also provides a chiral active pocket for both substrates and can exert stereoselectivity during the C–C coupling reaction (vide supra). The geometry of the organocatalyst was taken to examine the complex structures formed with isatin derived ketimines and 1-naphthol. The dual activation of substrate molecules can take place with the organocatalyst, where the Nu is activated through hydrogen bonding with tertiary nitrogen atom of alkaloid and the EI is activated through hydrogen bonding with thiourea –N–H groups.17,28 We have optimized the complex structures formed with the catalyst, isatin derived N-Boc ketimines and 1-naphthol. The complex structure optimized for both the possible attack (Re and Si face attack on N-Boc ketimines of 1-naphthol).
 | ||
| Fig. 7 Optimized structure of quinine thiourea organo catalyst calculated with B3LYP/6-31G(d) level of theory. The hydrogen bonding distance exist between tertiary nitrogen and Ha atom of thiourea is given in Å. (White: hydrogen, grey: carbon, blue: nitrogen, red: oxygen, cyan: fluorine, yellow: sulphur.) | ||
The optimized complex structures adopt a very similar arrangement of the Nu and EI with the organocatalyst as obtained in the previous studies.17,29 The 1H and 13C NMR studies performed corroborate the complex structures.
The 1H NMR studies suggest that the –NHb proton of organocatalyst shows more downfield shift compared to the –NHa proton of catalyst in presence of N1-protected isatin ketimine, however the nucleophile (1-naphthol) is hydrogen bonded with the tertiary nitrogen atom of the organocatalyst. The DFT analysis of complex structures formed among the 1-naphthol and N1-benzylisatin derived N-Boc ketimine with chiral organocatalyst also suggest that the carbonyl oxygen of isatin ketimine is strongly hydrogen bonded with –NHb proton of the catalyst compared to –NHa proton in both the possible pathway (distances are 1.86 Å, 2.77 Å in Re face and 1.82 Å, 1.93 Å in Si face attack respectively, Scheme 1, Fig. 8). Whereas, the nucleophile is stabilized via hydrogen bonding interaction exist between the enolic –OH group of naphthol and the tertiary nitrogen atom of quinine alkaloid (1.11 Å and 1.10 Å for Re and Si face respectively, Fig. 8). The NBO charges calculated for C-6 and C-8 in the catalyst are −0.174 and −0.019, respectively. In the Re face complex, the NBO charge on C-6 (−0.169) and C-8 (0.00) is smaller than the catalyst (ESI, Fig. S1†). Therefore, the NBO charge calculations suggest that the C-6 and C-8 carbon atoms of the catalyst in Re face complex experience down field shift in 13C NMR spectroscopy. The C-6 and C-8 carbon atoms in the complex structure is more electropositive compared to the catalyst. Therefore, the hydrogen bonding interactions are in the line of good agreement with the observed 1H and 13C NMR spectral results (Fig. 8 and S1†).
 | ||
| Fig. 8 Complex structures of 1-naphthol and N1-benzylisatin derived ketimine as with chiral quinine thiourea calculated with B3LYP/6-31G(d) level of theory. The distances are given in Å. (White: hydrogen, grey: carbon, blue: nitrogen, red: oxygen, cyan: fluorine, yellow: sulphur.) | ||
Nonetheless, there is another pathway to achieve the asymmetric Friedel–Craft reaction products with quinine thiourea organocatalyst and 1-naphthol and polarized carboxylate carbon of N1-protected isatin ketimines. In this pathway, the Nu can be activated by the N-Ha and N-Hb protons of the thiourea unit of the catalyst and EI can attach to the N–H of the protonated amine (II) (Fig. S2 and S3†). The M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) calculated results show that the Re and Si face complex is energetically unstable compared to the corresponding Re and Si face complex observed in the former case (I) (Fig. S2 and S3†).
The DFT calculations and NMR studies showed the appropriate arrangement of substrates with the catalyst, the transition states have been located at B3LYP/6-31G(d) level of theory and shown in Fig. 9. We have determined the transition states for the attack of 1-naphthol to the Re and Si face of N1-benzyl isatin derived ketimine with the organocatalyst complexed with both the substrate molecules. The single point energy calculations have been performed at M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory for these transition states and the relative energies are given in Fig. 9.
 | ||
| Fig. 9 Transition state geometries of 1-naphthol and N1-benzylisatin derived ketimine as with chiral quinine thiourea organocatalyst towards the Friedel–Craft reaction calculated at B3LYP/6-31G(d) level of theory. Single point energy calculated at M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory by using SCRF-SMD framework for dichloromethane continuum. The distances are given in Å. | ||
The calculated results show that the Re face attack is 4.4 kcal mol−1 more stable than the corresponding Si face attack of 1-naphthol to N1-benzylisatin derived ketimine (Fig. 9). The solvent phase calculated results also suggest that Re face attack predominates over Si face attack by 3.3 kcal mol−1 (Fig. 9). The Gibb's free energy calculated results for these transition states further confirmed the preference in Re face attack over Si face attack (Fig. 9). The computed results reveal that the enantiomeric excess in asymmetric Friedel–Craft reaction of 1-naphthol to N1-benzyl isatin derived ketimine with Cat. is due to the preference of Re face approach. The recent experimental results suggest that 99% enantiomeric excess was observed for the N1-benzyl isatin derived ketimine with 1-naphthol in presence of quinine thiourea organocatalyst.16
The recently reported results on the formation of enantiomeric excess with isatin derived N-Boc ketimine and 1-naphthol in presence of quinine thiourea organocatalyst also showed that Re face approach is preferred over the Si face approach using the X-ray crystal structure analysis.16 Further, the efficiency of organocatalyst with N1-phenyl and N1-methyl isatin derived N-Boc ketimine has also been examined with DFT calculations. The transition states for the Re face and Si face attack of 1-naphthol on N1-phenylisatin derived ketimine have been computed at the same level of theory (Fig. 10). The calculated results show that the Re face attack of 1-naphthol to N1-phenylisatin derived ketimine is energetically favoured by 4.3 kcal mol−1 and in the dichloromethane is 3.7 kcal mol−1 (Fig. 10). The Gibb's free energy results also corroborate the experimental results.
 | ||
| Fig. 10 Transition state geometries of 1-naphthol and N-phenylisatin with chiral quinine thiourea organocatalyst towards the Friedel–Craft reaction calculated at B3LYP/6-31G(d) level of theory. Single point energy calculated at M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory by using SCRF-SMD framework for dichloromethane continuum. The distances are given in Å. | ||
The study was extended with N1-methylisatin derived ketimine as an electrophile for this asymmetric Friedel–Craft reaction. The computed results predicted the favored Re-face approach compared to the Si-face approach of 1-naphthol to N1-methyl isatin derived ketimine (Fig. 11).
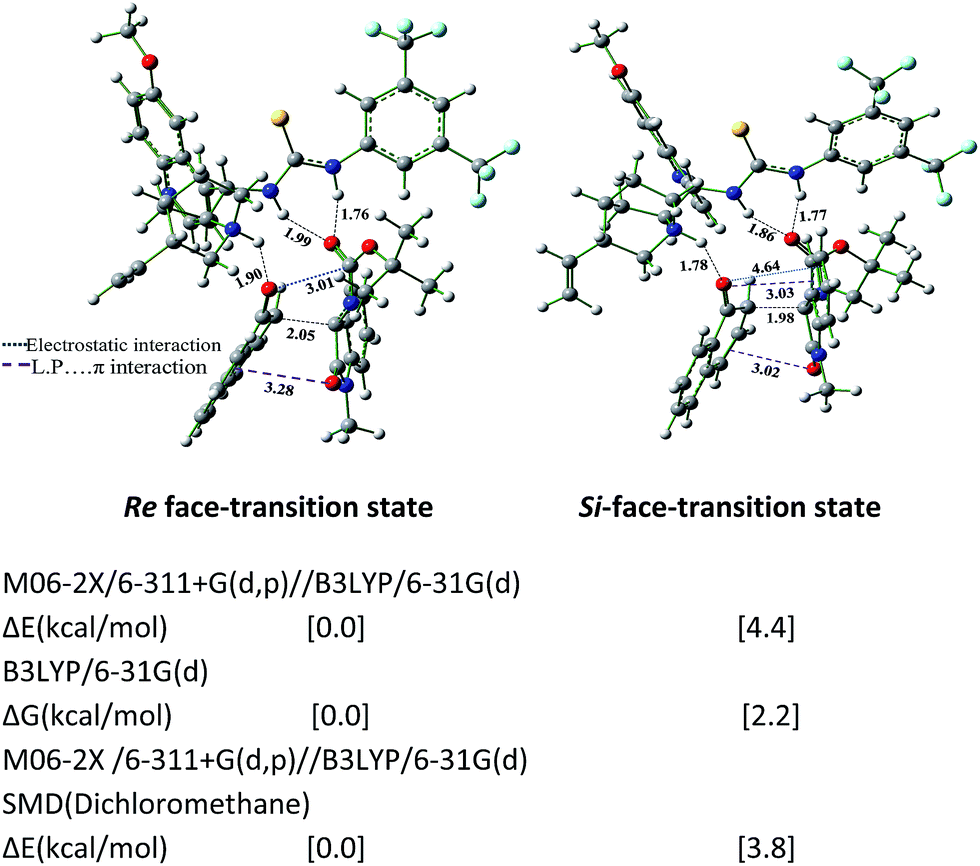 | ||
| Fig. 11 Transition state geometries of 1-naphthol and N-methylisatin with chiral quinine thiourea organocatalyst towards the Friedel–Craft reaction calculated at B3LYP/6-31G(d) level of theory. Single point energy calculated at M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory by using SCRF-SMD framework for dichloromethane continuum. The distances are given in Å. | ||
These DFT calculated results are in good agreement with the observed experimental results.
The results reveal that the Re face attack would be favored over the Si face attack for all the systems in asymmetric Friedel–Craft reaction with quinine thiourea organocatalyst. The experimentally observed enantiomeric excess in such cases are in good agreement with the computed results.
The transition state structure analysis reveals that the favoured electrostatic attraction is present between the enolate oxygen of 1-naphthol and polarized carboxylate carbon of N1-protected isatin ketimines in the Re face transition states. The attractive interactions fall within the van der Waals distance of carbon oxygen interactions. However, in the case of Si face transition state, disfavoured interactions are present between oxygen lone pair (L.P) and pi (π) plane of the aromatic rings (Fig. 9–11). The N1-protected isatin ketimines shifted in the Si face transition state, hence experience the oxygen lone pair (L.P) and pi (π) plane interactions presumably responsible for the destabilization in this case. The calculated distance between the oxygen atom and the π-plane of the naphthol ring is ∼3.0 Å and can cause deleterious destabilization in the transition state geometry of the Si face attack. Similar interaction was also noticed for the Si face attack with 1-naphthol (π) plane and the carbonyl oxygen of N1-protected isatin ketimines (∼3.0 Å). The attractive electrostatic interaction in the Re face transition state and the disfavoured lone pair (L.P) and pi (π) interaction leads to larger energy difference and hence can yield such high enantiomeric excess in all cases.
Experimental
General method
All the isatins, naphthol were purchased and used as received. The solvents were dried and stored over activated molecular sieve under nitrogen atmosphere. 1H NMR and 13C NMR spectra of all the compounds were obtained from Bruker-Avance-DPX-200 (200 MHz) or 500 MHz spectrometer at ambient temperature using TMS as internal standard. The enantiomeric excess of chiral compounds were determined by chiral Shimadzu-HPLC with SPD-M10A-VP and SPD-M20A UV detector and PDR-Chiral Inc. advanced Laser Polarimeter (PDR-CLALP), using chiral Daicel Chiralpak columns with 2-propanol/hexane mixture as eluent of the reaction mixture. Absolute configurations of chiral compounds were determined by comparing the sign of optical rotation (obtained from PDR-CLALP) and comparison with the literature.Typical experimental procedure for the enantioselective aza-Friedel–Crafts reaction
To a solution of quinine thiourea 5b (5 mol%) in 2 ml of dichloromethane 1-naphthol (3 mmol), ketimine (2 mmol) and 50 mg of 4 Å MS were added under N2 atm. The reactions was monitored by TLC until the consumption of ketimine and after the completion of the reaction, solvent was removed under reduced pressure and it was subjected to column chromatography using hexane:ethyl acetate (9.5:5) as eluent to afford desired product 8 in pure form.
Conclusions
We successively tried to report the most efficient bi-functional cinchona thiourea system for the reaction of 1-naphthol with isatin derived N-Boc ketimine derivatives to form the most valuable Betti base derivatives in very good yields (99%) and excellent enantioselectivity (95–99%). Based on the kinetics, NMR experiments and DFT studies we have unraveled the origin of stereoselectivity using bi-functional cinchona thiourea catalyst for the reaction of 1-naphthol with isatin derived N-Boc ketimine derivatives. The computed results suggest that the attractive electrostatic interactions in the Re face transition state and the deleterious lone-pair⋯π interactions in the Si face transition state governed the formation of Re-face product in this aza-Friedel–Crafts reaction. This work also showcased the cooperation of multiple attractive non-covalent interactions to stabilize the transition state leading to the major enantiomer and thus a key component for enantioinduction.Acknowledgements
CSIR-CSMCRI registration no is 99/2015. Authors are thankful to UGC and Indus Magic Project CSC0123 for financial support. S. B. is thankful to CSIR for awarding JRF fellowship and AcSIR for Ph.D enrolment. Prathibha kumari is thankful to AcSIR for Ph.D enrolment. Analytical Discipline and Centralized Instrumental Facility is gratefully acknowledged for providing instrumental facilities.References
- (a) Friedel–Crafts and Related Reactions, ed. G. A. Olah, Wiley-Interscience, New York, 1963-65, vol. 1–4 Search PubMed; (b) G. A. Olah, R. Krishnamurti and G. K. S. Prakash, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 3, p. 293 Search PubMed; (c) M. Bandini and A. Umani-Ronchi, Catalytic Asymmetric Friedel–Crafts Alkylations, Wiley-VCH, Weinheim, 2009 Search PubMed; (d) T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS PubMed; (e) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS PubMed; (f) M. Bandini, A. Melloni, S. Tommasi and A. Umani-Ronchi, Synlett, 2005, 1199 CrossRef CAS; (g) K. A. Jørgensen, Synthesis, 2003, 1117 CrossRef.
- M. Betti, Org. Synth., 1941, 1, 381–384 Search PubMed.
- C. Cardellicchio, M. A. M. Capozzi and F. Naso, Tetrahedron: Asymmetry, 2010, 21, 507 CrossRef CAS PubMed.
- B. P. Mathew, A. Kumar, S. Sharma, P. K. Shukla and M. Nath, Eur. J. Med. Chem., 2010, 45, 1502 CrossRef CAS PubMed.
- M. Salamone, R. Amorati, S. Menichetti, C. Viglianisi and M. Bietti, J. Org. Chem., 2014, 79, 6196 CrossRef CAS PubMed.
- P. F. Kaiser, J. M. White and C. A. Hutton, J. Am. Chem. Soc., 2008, 130, 16450 CrossRef CAS PubMed.
- G. L. Cheng, X. Y. Wang, R. Zhu, C. W. Shao, J. M. Xu and Y. F. Hu, J. Org. Chem., 2011, 76, 2694 CrossRef CAS PubMed.
- X. Wang, Y. Dong, J. Sun, X. Xu, R. li and Y. Hu, J. Org. Chem., 2005, 70, 1897 CrossRef CAS PubMed.
- (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (b) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209 CrossRef CAS PubMed; (c) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 (Angew. Chem., 2007, 119, 8902–8912) CrossRef CAS PubMed; (d) F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS PubMed; (e) J. E. M. N. Klein and R. J. K. Taylor, Eur. J. Org. Chem., 2011, 6821 CrossRef CAS PubMed; (f) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327 RSC.
- P. Chauhan and S. S. Chimni, Tetrahedron: Asymmetry, 2013, 24, 343 CrossRef CAS PubMed.
- (a) N. Hara, S. Nakamura, M. Sano, R. Tamura, Y. Funahashi and N. Shibata, Chem.–Eur. J., 2012, 18, 9276 CrossRef CAS PubMed; (b) W. Yan, D. Wang, J. Feng, P. Li, D. Zhao and R. Wang, Org. Lett., 2012, 14, 2512 CrossRef CAS PubMed; (c) T.-Z. Li, X.-B. Wang, F. Sha and X.-Y. Wu, J. Org. Chem., 2014, 79, 4332 CrossRef CAS PubMed; (d) X.-B. Wang, T.-Z. Li, F. Sha and X.-Y. Wu, Eur. J. Org. Chem., 2014, 739 CrossRef PubMed; (e) J. Zhao, B. Fang, W. Luo, X. Hao, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 241 (Angew. Chem., 2015, 127, 243) CrossRef CAS PubMed.
- (a) H. Lv, B. Tiwari, J. M. Mo, C. Xing and Y. R. Chi, Org. Lett., 2012, 14, 5412 CrossRef CAS PubMed; (b) F.-L. Hu, Y. Wei, M. Shi, S. Pindi and G. Li, Org. Biomol. Chem., 2013, 11, 1921 RSC; (c) S. Nakamura, K. Hyodo, M. Nakamura, D. Nakane and H. Masuda, Chem.–Eur. J., 2013, 19, 7304 CrossRef CAS PubMed; (d) J. Xu, C. Mou, T. Zhu, B.-A. Song and Y. R. Chi, Org. Lett., 2014, 16, 3272 CrossRef CAS PubMed; (e) J. George, B. Sridhar and B. V. S. Reddy, Org. Biomol. Chem., 2014, 12, 1595 RSC.
- (a) T. Arai, E. Matsumura and H. Masu, Org. Lett., 2014, 16, 2768 CrossRef CAS PubMed; (b) M. Holmquist, G. Blay and J. R. Pedro, Chem. Commun., 2014, 50, 9309 RSC; (c) A. Kumar, J. Kaur, S. S. Chimni and A. K. Jassal, RSC Adv., 2014, 4, 24816 RSC; (d) Y.-H. Wang, Y.-L. Liu, Z.-Y. Cao and J. Zhou, Asian J. Org. Chem., 2014, 3, 429 CrossRef CAS PubMed.
- (a) Y.-L. Liu, F. Zhou, J.-J. Cao, C.-B. Ji, M. Ding and J. Zhou, Org. Biomol. Chem., 2010, 8, 3847 RSC; (b) D. Wang, J. Liang, J. Feng, K. Wang, Q. Sun, L. Zhao, D. Li, W. Yan and R. Wang, Adv. Synth. Catal., 2013, 355, 548 CAS; (c) Y.-L. Liu and J. Zhou, Chem. Commun., 2013, 49, 4421 RSC.
- J. Feng, W. Yan, D. Wang, P. Li, Q. Sun and R. Wang, Chem. Commun., 2012, 48, 8003 RSC.
- M. Montesinos-Magraner, C. Vila, R. Canton, G. Blay, I. Fernandez, M. C. Munoz and J. R. Pedro, Angew. Chem., Int. Ed., 2015, 54, 6320 CrossRef CAS PubMed.
- J.-L. Zhu, Y. Zhang, C. Liu, A.-M. Zheng and W. Wang, J. Org. Chem., 2012, 77, 9813 CrossRef CAS PubMed.
- For some recent books, see: (a) A. Berkessel and H. Gröger, Asymmetric Organocatalysis, Wiley Online Library, 2005 Search PubMed; (b) B. List, Asymmetric Organocatalysis, Springer, New York, 2010 Search PubMed; (c) H. Pellissier, Recent Developments in Asymmetric Organocatalysis, RSC Publishing, Cambridge, 2010 Search PubMed.
- (a) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Celebi-Olcum and K. N. Houk, Chem. Rev., 2011, 111, 5042 CrossRef CAS PubMed; (b) K. N. Houk and P. H.-Y. Cheong, Nature, 2008, 455, 309 CrossRef CAS PubMed.
- (a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed; (b) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed.
- (a) A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227 CrossRef CAS; (b) U. C. Singh and P. A. Kollman, Comput. Chem., 1986, 7, 718 CrossRef CAS PubMed; (c) M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700 CrossRef CAS PubMed; (d) J. Aqvist and A. Warshel, Chem. Rev., 1993, 93, 2523 CrossRef; (e) S. R. Billeter, S. P. Webb, T. Iordanov, P. K. Agarwal and S. Hammes-Schiffer, J. Chem. Phys., 2001, 114, 6925 CrossRef CAS PubMed.
- (a) S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 1, 461 Search PubMed; (b) T. Vreven and K. J. Morokuma, Comput. Chem., 2000, 21, 1419 CrossRef CAS; (c) K. Morokuma, Philos. Trans. R. Soc. London, Ser. A, 2002, 360, 1149 CrossRef CAS PubMed; (d) T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery Jr, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815 CrossRef CAS; (e) A. Sengupta and R. B. Sunoj, J. Org. Chem., 2012, 77, 10525 CrossRef CAS PubMed.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed; (b) J.-S. Oh, J. W. Lee, T. H. Ryu, J. H. Lee and C. E. Song, Org. Biomol. Chem., 2012, 10, 1052 RSC.
- (a) N. Agmon, J. Phys. Chem. B, 2007, 111, 7870 CrossRef CAS PubMed; (b) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed; (c) M. J. Climent, A. Corma, I. Domínguez, S. Iborra, M. J. Sabater and G. Sastre, J. Catal., 2007, 246, 136 CrossRef CAS PubMed.
- (a) A. Moran, A. Hamilton, C. Bo and P. Melchiorre, J. Am. Chem. Soc., 2013, 135, 9091 CrossRef CAS PubMed; (b) A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS; (c) S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12795e |
| This journal is © The Royal Society of Chemistry 2015 |