
Porous silicon nanoparticles as a nanophotocathode for photoelectrochemical water splitting†
Abstract
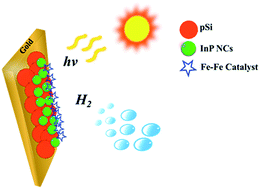
The antireflective properties and nanometer sizes of silicon nanoparticles can be exploited for improved solar energy conversion. We report on using porous silicon nanoparticles as a photocathode for photoelectrochemical water splitting. An enhancement in the photocurrent density was observed when porous silicon nanoparticles were decorated with indium phosphide nanocrystals and a bio-inspired iron sulfur carbonyl electrocatalyst. Our system gave a photocurrent density of −2.2 μA cm−2 while generating hydrogen gas.
 Please wait while we load your content...
Please wait while we load your content...

