
Combined experimental and theoretical insight into the drug delivery of nanoporous metal–organic frameworks†
Abstract
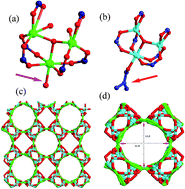
Two isostructural nanoporous MOFs with [Zn3(μ3-O)(BTC)2(H3O)]n (NTU-Z11) and {[Zn3(μ3-O)(BTC)2(DMF)]·2NH2(CH3)2·4H2O}n (GDMU) (BTC = 1,3,5-benzenetricarboxylate) have been used as drug carriers of 5-fluorouracil (5-FU). The incorporation of 5-FU into the desolvated NTU-Z11 and GDMU was around 0.38 g g−1 and 0.22 g g−1, respectively. NTU-Z11 presents pH-triggered controlled drug release properties in pH 6.0, 7.4 and 9.18 and water media. In addition, we performed GCMC simulations to investigate the loading of 5-FU into NTU-Z11 and GDMU at the molecular level. The results from simulations reproduce the experimental trend with respect to the drug loading capacity of each material. A comparison between the calculated drug loading values and some molecular level properties indicates the existence of a relationship between the void space of the material and the drug loading capacity.
 Please wait while we load your content...
Please wait while we load your content...

