
A DFT study of the chemical and optical properties of 7-atom Ag–X [X = Li, Na] nanoalloys for potential applications in opto-electronics and catalysis
Shaikat Debnathab,
Suhana Mohd Said*a,
Franck Rabilloudc,
Abhijit Chatterjeed,
Muhammad Faris Roslana,
Azizah Mainale and
Mohamad Syafie Mahmoodaf
aDepartment of Electrical Engineering, Faculty of Engineering, University of Malaya, Kuala Lumpur 50603, Malaysia. E-mail: smsaid@um.edu.my
bBangladesh Power Development Board, Ministry of Power, Energy and Mineral Resources, Government of the People's Republic of Bangladesh, Bangladesh
cInstitut Lumière Matière, UMR5306 Université Claude Bernard Lyon 1-CNRS, Université de Lyon, 69622 Villeurbanne cedex, France
dMaterials Science, Accelrys K.K., Kasumiagaseki 3-7-1, Tokyo 100-0013, Japan
eDepartment of Chemistry, Faculty of Science, University of Malaya, Kuala Lumpur 50603, Malaysia
fFaculty of Applied Science, MARA University of Technology, Shah Alam 40450, Malaysia
First published on 28th October 2015
Abstract
In this paper, Ag atoms are substituted by X (Li, Na) atoms to form AgmX(7−m) clusters to explore their electronic, chemical and optical properties in the framework of density functional theory (DFT). The clusters are geometrically optimized without imposing symmetry and later, vibrational analysis is carried out to test the stability of the optimized structures. The calculation of ionization potential and electron affinity asserted that the Li and Na doped bimetallic clusters (especially, Ag4Li3 and Ag3Li4) are very stable in the neutral state, but their anions are expected to be very reactive. The calculated absorption spectra of the AgmX7−m clusters have revealed that the doping of Li and Na has made the absorption band wider with regards to undoped Ag7 clusters. Therefore, this work suggests that Li and Na doping (especially, Ag4X3, Ag3X4 and Ag2X5 clusters) will result in improvement of the absorption band in the 1–5 eV range, which is the prime absorption band for opto-electronic devices such as solar cells.
1. Introduction
Bimetallic nanoclusters, which are the aggregation of two types of metal atoms of nanoparticle size, have been utilized as key components in many catalytic, optical and magnetic devices. They have been subject to the interest of both theoretical and experimental researchers for their distinct physical and chemical properties.1 Presently, there is an growing interest in bimetallic nanoparticles, compared to monometallic nanoparticles because bimetallization can offer space for improvement in optical, catalytic and magnetic performance over the original pure mono-metal nanoparticles and can create new properties. Also, molecular tuning of these bimetallic nanoclusters may give rise to novel properties which are not realized in bulk or, monometallic nanoparticles.2,3 The properties of these bimetallic nanoclusters are not solely size-dependent, rather the chemical composition and molecular arrangement significantly affect their specific structural,4–7 electronic,5,7 optical4,8 and magnetic properties9 which has led to a technological interest in catalysis10,11 and also in the application and development of new nano-devices for electronics.2,12Among the hundreds of bimetallic nanoalloys explored till date, Ag–X [X = Li, Na] nanoalloys are of special interest because of the special chemical and optical feature of both silver and alkali metals nanoparticles. In research history of nanomaterials, silver is the most commonly used materials due to their comparatively low optical losses in the visible and near-infrared (NIR) region. For example, the usages of silver have been demonstrated as negative refractive index material, hyperlens,13 superlens14 and optical transmission.15 However, when the nano-fabrication is concerned, silver degrades comparatively quickly and the thickness threshold for uniform continuous films is around 12–13 nm, which is a major barrier for transformation optic (TO) and several other micro and nano-devices.16,17 In addition, silver's dependency on the roughness of the surface with regards to optical losses has also reduced its viability in applications.18 Apart from the technical disadvantages mentioned so far, the main demerits of silver is their relatively high prices and limitation in resources, which eventually render them unsuitable for commercial applications. On the other hand, amongst the nanometals reported to date for their plasmonic potential, lithium, sodium and potassium have shown to possess the highest absorption efficiency (Qabs), coupled with very low inter-band transition losses at optical frequencies.19,20 Of particular merit, these transition losses have been noted to be comparable or surpass that of silver and gold. This is coupled with the additional advantage of exhibiting the strongest free-electron-like-behavior, which result in very prominent surface plasmon resonance (SPR) in visible-UV range.18 The only drawback that had been restricting alkali metals to be used extensively in nanotechnology is their extreme reactive nature towards air and water. Therefore, alloy containing silver and alkali metals will allow us to use the excellent plasmonic properties of alkali metals, whereas silver atoms can work as the screening to capping the alkali metals to restrict their chemically reactive nature. Hence it is expected that this Ag–X nanoalloy can be an excellent improvement over the bare silver nanoparticles.
In order to exploring the chemical and optical properties of the Ag–X nanoalloys, we have picked up the decahedral shape for its special chemical and optical features. Decahedral is a special one being the intermediate building blocks for rods and wire shaped nanoparticles.21 Moreover, their remarkable optical properties has made decahedral silver nanoparticle a popular commercial product (such as SCIVENTION's 35–120 nm sized decahedral silver) for the opto-electronic, catalytic and biological applications.21,22 In our current work we have used the 7-atoms decahedral Ag7 structures with D5h symmetry as the quantum model, which is a reported global minimum geometry for 7 atoms silver, lithium and sodium as well.23,24 Later silver atoms have been replaced by the X atoms to form AgmX(7−m) nanoalloys. In our previous work25,26 the electronic, chemical and optical properties of Ag–X [X = Li, Na, K] nanoalloys were explored in the framework of density functional theory (DFT). In that work, single X atom was added by replacing an silver atom from the 13-atoms decahedral silver cluster; hence only 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for Ag:X was checked. In contrary, in our current work all the mixing ratios in between Ag:X = 6:1 to Ag:X = 1:6 are explored to find out the best fit alloying ratio. Apart from our previous reports, several articles on photoionization spectroscopy of metal dimers including Ag–Li,27,28 Ag–Na,29 Ag–K30 have been published till date but to our knowledge this is the first report on very small Ag–X bimetallic clusters of less than 1 nm diameter size, where a wide range of mixing ratios have been reported.
:1 ratio for Ag:X was checked. In contrary, in our current work all the mixing ratios in between Ag:X = 6:1 to Ag:X = 1:6 are explored to find out the best fit alloying ratio. Apart from our previous reports, several articles on photoionization spectroscopy of metal dimers including Ag–Li,27,28 Ag–Na,29 Ag–K30 have been published till date but to our knowledge this is the first report on very small Ag–X bimetallic clusters of less than 1 nm diameter size, where a wide range of mixing ratios have been reported.
In this paper, a systematic investigation of electronic and optical properties of doped neutral clusters of AgmX(7−m) is explored against its potential for opto-electronic and catalytic applications. For this purpose, m = 1–6 Ag atom has been replaced by an alkali metal atom (X = Li, Na) to form AgmX(7−m) bimetallic clusters. Using these geometrically optimized structures, DFT based calculations of binding energy, HOMO–LUMO gap; vertical ionization potential (VIP) and vertical electron affinity (VEA) are carried out to characterize the stability and chemical inertness of the doped bimetallic clusters. TDDFT calculation has been carried out to predict the optical spectra of the clusters, which is then followed by the partial density of states (PDOS) calculations.
In the following section, a brief description of the computational method will be provided. Then, the structural and electronic properties will be presented, followed by optical spectrum and PDOS of the clusters.
2. Computational method
In this study, the investigation of geometrical and energetic stabilities and optical properties of the doped AgmX(7−m) bimetallic structures are investigated using the density functional spin-polarized calculations using DMol3 Accelrys Inc. code.31,32 All geometrical structures of the neutral clusters are optimized without imposing any symmetry or geometry constrains. Calculations have been performed using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.33 The Kohn–Sham equation was expanded in a double numeric quality basis set (DNP) with polarization functions. The valence configurations of Ag, Li and Na are 4d105s1, 1s22s1 and 2p63s1, respectively; hence the multiplicity is 2 for all the neutral AgmX7−m clusters. To consider the relativistic effect, the semi-core pseudo-potentials34 are used for the treatment of the core electrons of the doped clusters. The orbital cut off range and Fermi smearing were selected as 5.0 Å and 0.001 Ha respectively. The self-consistent-field (SCF) procedures were performed with a convergence criterion of 10−6 a.u. The energy, maximum force and maximum displacement convergence were set as 10−6 Ha, 0.002 Ha Å−1 and 0.005 Å respectively. The ALDA kernal exchange co-relation method was employed with TDDFT for calculating the optical excitation spectra.35 The absorption spectra presented in the figures below give the oscillator strength as a function of the excitation energy, together with a curve obtained by Lorentzian broadening.3. Structural and energetic stability
The first aspect of investigation is to ensure the geometric and energetic stability of the Ag–X clusters. For the quantum model, a 7-atom decahedron cluster with D5h symmetry was selected, where silver atoms from Ag7 was replaced to form AgmX7−m bimetallic clusters. The variations are shown in Fig. 1. The decahedron with D5h symmetry has been identified as the lowest-energy structure for Ag7, Li7 and Na7 by other researchers. For alloys, we have only considered the decahedron structure, without any symmetry constraints, because we are interested in characterizing the changes in electronic, chemical, optical properties when Ag atoms are substituted by Li or Na atoms. Thus, the scope addressing the energy considerations specific to substitutions to the Ag atoms in the decahedral geometry. Fig. 1 and 2 shows all the doping positions and lowest energy isomers for 7 atoms AgmX7−m nanoclusters along with their respective binding energies (BE). Ag7, Li7 and Na7 clusters have the geometrical global minima with D5h symmetry with binding energies of 9.61 eV, 6.41 eV and 4.46 eV respectively which is in accordance with the previous reports.23–26 Among the same doped numbered groups, the structures with highest binding energies, i.e. panels b, d, j, p, s, and v in Fig. 1 and panels b, d, j, n, s and v in Fig. 2, are selected for further electronic, chemical and optical properties' calculations. It was instructive for us at this stage, to conduct confirmation of the stability for the pure and doped clusters through harmonic vibrational frequency calculations. For this procedure, the stability of the clusters was confirmed by the absence of imaginary frequencies. The lowest and highest vibrational frequencies of the clusters are charted in Table 1. | ||
| Fig. 1 Lowest-energy structures for 7-atoms AgmLi7−m clusters. Point group symmetry and binding energy are given. Li and Ag atoms are in red and green respectively. | ||
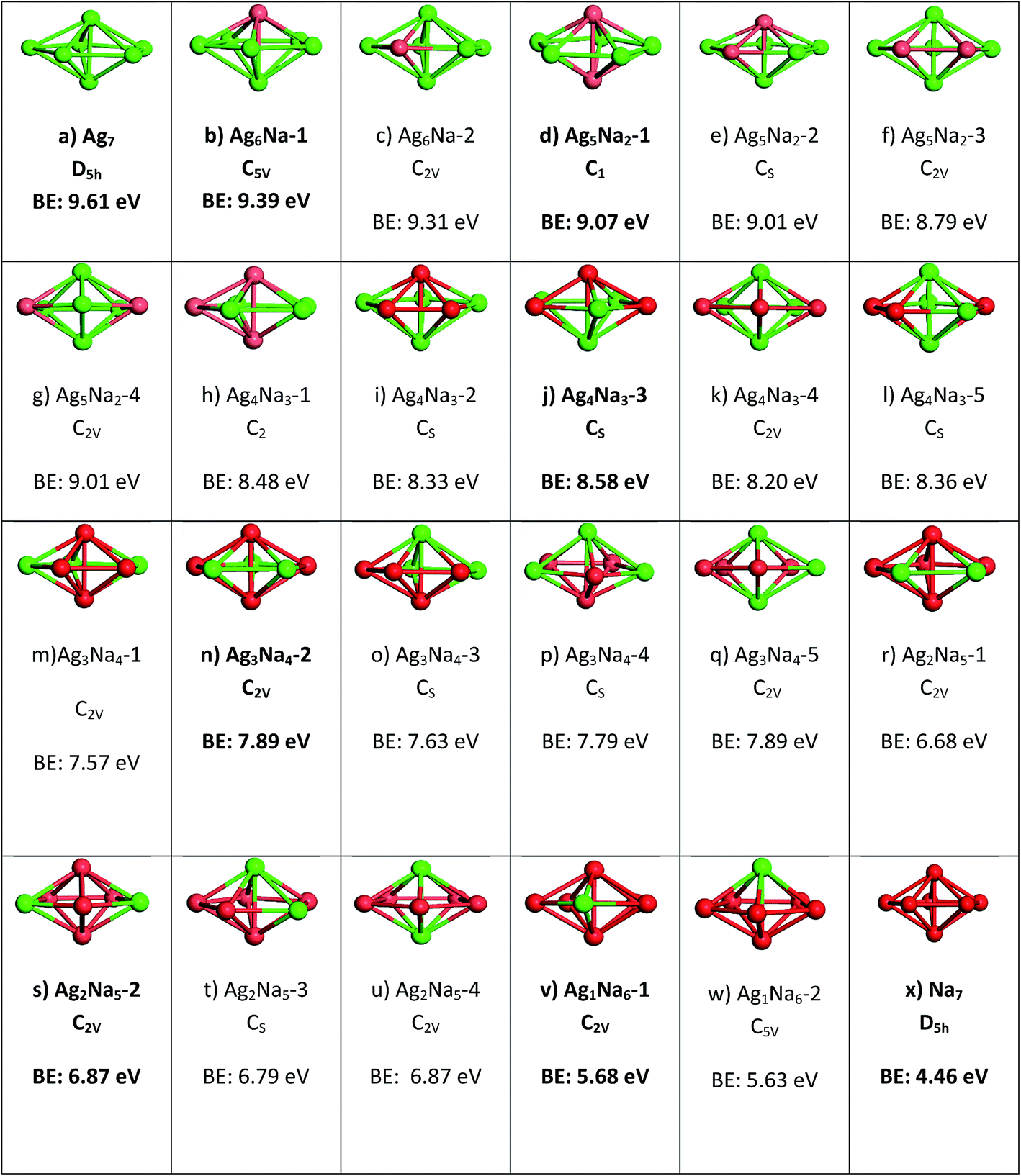 | ||
| Fig. 2 Lowest-energy structures for 7-atoms AgmNa7−m clusters. Point group symmetry and binding energy are given. Na and Ag atoms are in red and green respectively. | ||
| Compound | Lowest vibrational frequency cm−1 | Highest vibrational frequency cm−1 | Compound | Lowest vibrational frequency cm−1 | Highest vibrational frequency cm−1 |
|---|---|---|---|---|---|
| Ag7 | 40.41 | 158.24 | Ag7 | 40.41 | 158.24 |
| Ag6Li1 | 26.67 | 334.64 | Ag6Na1 | 29.03 | 156.53 |
| Ag5Li2 | 29.13 | 351.05 | Ag5Na2 | 23.42 | 182.25 |
| Ag4Li3 | 39.37 | 346.62 | Ag4Na3 | 45.20 | 186.80 |
| Ag3Li4 | 44.76 | 327.05 | Ag3Na4 | 41.03 | 169.52 |
| Ag2Li5 | 63.65 | 332.56 | Ag2Na5 | 45.90 | 163.90 |
| Ag1Li6 | 83.70 | 350.81 | Ag1Na6 | 47.85 | 155.89 |
| Li7 | 60.58 | 313.52 | Na7 | 33.87 | 176.62 |
4. Electronic and chemical properties
After discussing the geometrical structure and stability of the doped clusters in the previous section, now we focus our attention towards the electronic structure of these doped clusters and for that Tables 2 and 3 are compiled with binding energy (BE), ionization potential (IP), electron affinity (EA) and HOMO–LUMO gaps of the entire AgmX7−m nanoclusters. In times of calculating IP and EA, the ionized clusters are of same geometry of the neutral clusters; hence the IP and EA of the clusters are actually vertical ionization potential and vertical electron affinity. At this point, before moving towards their electronic and chemical properties, let's first make sure that our functional basis sets have calculated the data perfectly. From the data provided in Tables 2 and 3, it is evident that Ag7 is showing binding energy, VIP, VEA and HOMO–LUMO gap of 9.61, 6.43, 1.34 and 0.34 eV respectively, which is a fine match with previous reports.24,36–38 Similarly, the electronic and chemical parameters of Li and Na charted in the table are found to be agreed with the previous reports.39–42| Compounds | Binding energy | Vertical ionization potential | Vertical electron affinity | HOMO–LUMO gap |
|---|---|---|---|---|
| Ag7 | 9.61 | 6.43 | 1.34 | 0.34 |
| Ag6Li1 | 10.00 | 6.23 | 1.13 | 0.34 |
| Ag5Li2 | 10.17 | 5.94 | 1.05 | 0.35 |
| Ag4Li3 | 10.22 | 5.93 | 0.79 | 0.45 |
| Ag3Li4 | 9.96 | 5.79 | 0.30 | 0.82 |
| Ag2Li5 | 8.84 | 4.72 | 0.23 | 0.53 |
| Ag1Li6 | 7.64 | 4.53 | 0.18 | 0.56 |
| Li7 | 6.41 | 4.36 | 0.16 | 0.58 |
| Compounds | Binding energy | Vertical ionization potential | Vertical electron affinity | HOMO–LUMO gap |
|---|---|---|---|---|
| Ag7 | 9.61 | 6.43 | 1.34 | 0.34 |
| Ag6Na1 | 9.39 | 5.39 | 1.29 | 0.32 |
| Ag5Na2 | 8.79 | 5.37 | 1.11 | 0.36 |
| Ag4Na3 | 8.58 | 4.83 | 0.81 | 0.48 |
| Ag3Na4 | 7.89 | 4.28 | 0.48 | 0.46 |
| Ag2Na5 | 6.87 | 4.16 | 0.43 | 0.41 |
| Ag1Na6 | 5.68 | 4.02 | 0.36 | 0.34 |
| Na7 | 4.46 | 3.39 | 0.31 | 0.31 |
As a first step towards the confirmation of the electronic stability of the nanoclusters, the average binding energy per atom is calculated in the following way, where E refers to the energy of the attached cluster and X = Li and Na.
| BE (AgmX7−m) = mE (Ag) + (7 − m)E (X) − E (AgmX7−m) |
From the data presented in Table 2, it is evident that the AgxLi7−x clusters maintain an interesting pattern of binding energies. With respect to Ag7 cluster, the binding energy increases with the addition of Li into the system. More interestingly, it keeps increasing with the number of Li atoms doped into the system and reaches to its highest value with Ag4Li3 by maintaining the following pattern: Ag7 < Ag6Li1 < Ag5Li2 < Ag4Li3. From the next alloy, i.e. from Ag3Li4 to Li7, decrementing pattern is observed with Li clusters having the lowest binding energy amongst all the nanoclusters. The gap in the Pauling electronegativity of Ag (1.93) and Li (0.98) can explain this result. The large gap of electronegativity between Ag and Li have most probably created some ionic bonding and this ionic bonding gets stronger with equal number of atoms from both of Ag and Li. Hence, the binding energies of all the clusters in between Ag6Li1 to Ag3Li4 are higher with respect to Ag7, while the highest binding energy is observed in Ag4Li3.
On the contrary, very opposite feature is observed in Na-doped clusters, where the binding energy followed a decreasing graph from Ag6Na1 to Ag1Na6. Moreover, all the doped clusters have lower binding energy than Ag7. As like as Li-doped clusters, the Ag–Na alloys were also expected to follow a similar pattern. But the atomic size of Ag and Na has played the key role here. The atomic radii of Ag (165 pm) and Li (167 pm) are very close; hence doping Li atoms by replacing Ag atoms doesn't harm the geometrical harmony. But the atomic radii of Na (190 pm) is quite higher than Ag; therefore doping far bigger Na atoms by replacing Ag seriously dispatch the geometry of the Ag–Na alloys which is liable for their low binding energies.
Subsequently, our attention is then focused towards the VIP and VEA values, which are key parameters needed in the analysis of the chemical nature of small clusters. Given our knowledge that a higher value of IP specifies a deeper HOMO energy level, which in turn signifying that the structures with large value of IP are expected to be chemically more stable or, exhibiting lesser chemical reactivity. On the contrary, higher value of EA indicates a more solid binding between the cluster and an electron. Thus, low EA and IP values imply more chemically reactive nature of the clusters. Here, from the data provided in Tables 2 and 3, it is clear that IP and EA follow a declining trend from Ag7 to Ag1X6 clusters. It is well known that in the periodic table, the ionization energy decreases from top to bottom and increases from left to right. Thus, both of Li and Na possess lower EA and IP with respect to Ag; hence addition of alkali atoms into Ag will make the alloy' IP and EA lower than the pure one. Thus, the alloyed clusters have lower IP and EA compared to the pure Ag clusters, which in turn implies that the Ag–X alloyed clusters are chemically more reactive than the Ag clusters. In addition, it needs to be mentioned that the vertical electron affinity of AgmLi7−m follows a declining trend with increasing m, while the HOMO–LUMO gap is found to be increase with m (Table 2). The variations are monotonic, and reflect the differences between Ag and Li. The VEA of Ag7 and Li7 are 1.34 and 0.16 eV respectively, thus the decrease in VEA with increasing m is expected. Similar comments can be made for Na. At the atomic level, the EA of alkali atoms is about half that of silver. As a matter of fact, the EA given in Table 2 is the VEA, i.e. the anion is calculated at the geometry of the neutral specie, so a low vertical EA cannot be considered as an indicator of a low stability of the anion.
The energy gap between the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) is considered as an important quantity for evaluating the kinetic and chemical stability of the clusters. A signature feature of clusters with large HOMO–LUMO gaps is their thermodynamic and chemical stability. This is because it is energetically unfavorable to add electrons to a high-lying LUMO, to extract electrons from a low-lying HOMO, and so to form the activated complex of any potential reaction.43,44 In this point, looking at the numbers provided in Tables 2 and 3, it is apparent that Li7 possesses better chemical stability with larger HOMO–LUMO gap compared to Ag7 and Na7, whereas Na7 is chemically least stable amongst. Amongst all the Ag–X bimetallic clusters, Ag3Li4 retains the highest band gap with 0.82 eV; hence expected to be the most chemically stable amongst all. The probable reason behind its high band-gap is the presence of ionic bonding because of the large gap of electronegativity between Ag and Li. When we are looking the trend of HOMO–LUMO gap of the doped clusters, it is clear that the doped clusters' band gaps get higher from Ag7 to Ag3X4, reaches its highest value in Ag3Li4 and Ag4Na3 and then again starts to fall down. Moreover, the band gap values are higher for Ag–Li clusters with respect to Ag–Na clusters, which is because Li7 structures own high HOMO–LUMO gaps.
Given the current discussions on the electronic attributes of the AgmX7−m clusters, it is strongly implied that the doped clusters retained excellent electronic and chemical properties. In particular, the neutral clusters with high HOMO–LUMO gaps are expected to be very reactive as cations. This is due to their lower EA values as it becomes easier to lose electrons from the orbital, given the lower LUMO values.45 Similarly, in this work it is evident that the AgmX7−m clusters exhibit high HOMO–LUMO gap with low electron affinity. Especially, Ag3Li4 and Ag2Li5 clusters possess high HOMO–LUMO gaps with very small electron affinity; hence they should be excellent potential for catalytic applications.
5. Optical spectra and partial density of states (PDOS)
The absorption spectra of doped clusters are shown in Fig. 2 and 3. The oscillator strength is provided as a function of the excitation energy. In each case, Lorentzian broadening (with a full width at half-height of 0.1 eV) is applied to exclude the oscillations from the excited electrons and in order to provide a result comparable to the experimental results. As the peak optical spectra of both of Ag and X (Li, Na) lie in between 2 and 5 eV, these optical spectra are limited up to 5 eV as well. For instance, in our calculation the primary optical peak of Ag, Li and Na are 3.74 eV, 2.75 eV and 3 eV respectively, which are fine match to the previous reports.46–49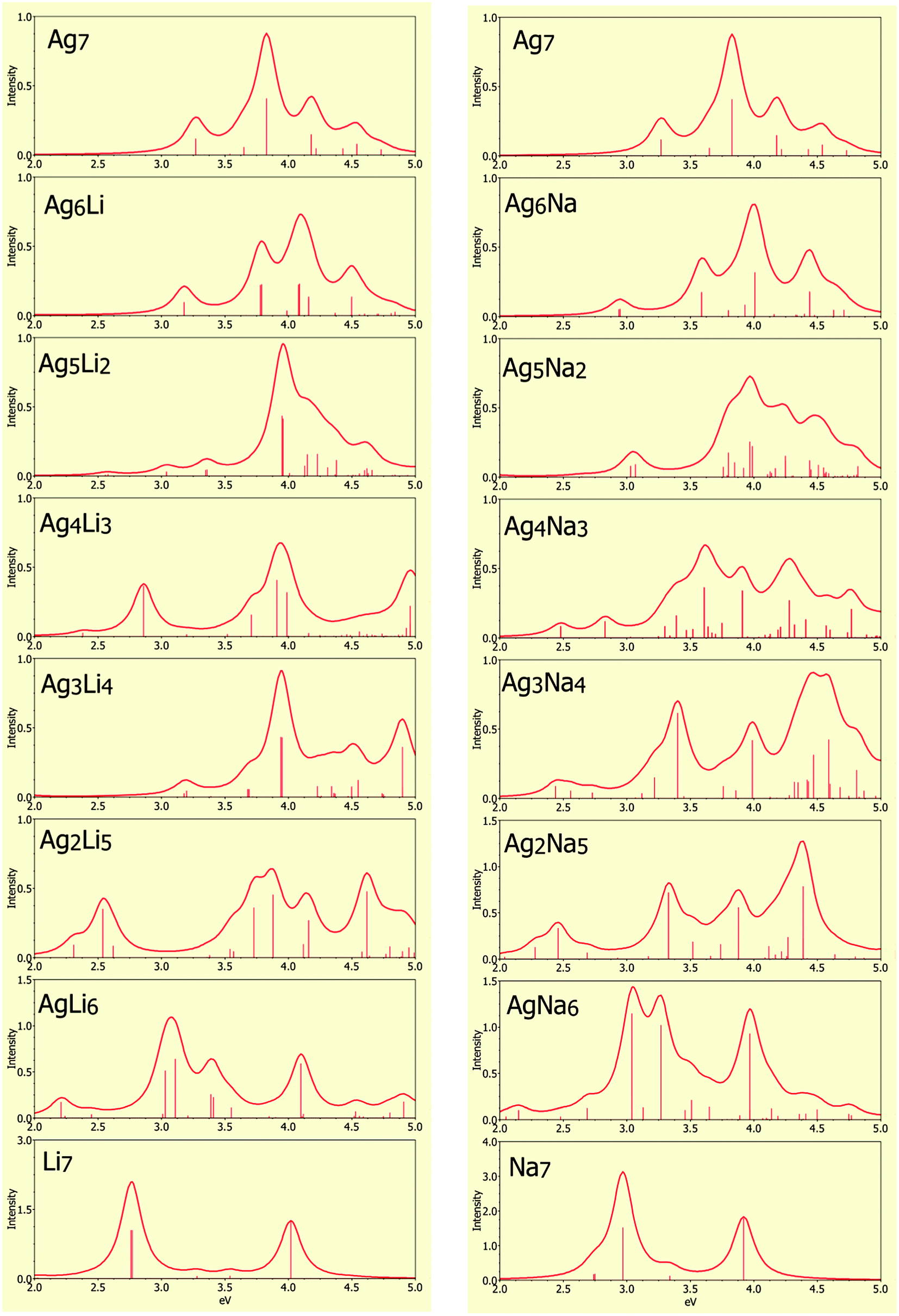 | ||
| Fig. 3 Optical absorption spectra of AgmX7−m bimetallic clusters. | ||
The optical spectrum of Ag is characterized by an intense and narrow band centered at 3.74 eV, whereas the Li doping into Ag has yielded some interesting spectra. In the spectrum of Ag6Li and Ag5Li2, the peak is slightly blue-shifted whereas the optical intensity is found to be marginally increased with respect to Ag7. More interesting spectrum is yielded by Ag4Li3, where the spectra got broadened with more peaks in lower frequencies. Similar broad-spectra are found for Ag2Li5 and Ag1Li6 clusters as well. But the highest peak is blue-shifted with respect to Ag7 for the Ag1Li6 cluster when this cluster offer higher light absorption intensity compared to all other AgmLi7−m clusters.
As like as Li-doped clusters, Na doping has also produced interesting changes in optical spectra. The absorption peak of Ag6Na and Ag5Na2 are marginally blue-shifted, when Ag5Na2 has offered more broadened spectrum with respect to Ag7. The optical spectra of Ag4Na3, Ag3Na4 and Ag2Na5 clusters seem to be more interesting as they offer large absorption spectra with higher intensity from 2 to 5 eV, while their peaks are blue-shifted with respect to Ag7. The spectrum of Ag1Na6 is similar to Na7, while Ag1Na6 possesses more absorption peaks compared to Na7. However, the sodium cluster here offers more absorption intensity.
Now let's move our focus towards the electronic evolution of the spectra of the doped clusters. Here, with the presence of alkali atoms these bimetallic AgmX7−m clusters in the excited states lead to a quasi-continuum spectrum. In particular, for the case of Ag7, the electronic transitions are happening as a result of excitations from the s and d orbitals. These s and d orbitals are well localized in surface silver atoms, along with its evolution as hybrid s–p orbitals in the wide areas of outer region where several atoms are involved with the system.50 On the contrary, the absorption spectra of alkali metal clusters are explained as collective oscillation of s valence electrons and the shift of absorption energies of very small particles, compared to the larger ones explained by a spill out (extension of the electronic wave functions out of the “classical volume” of the cluster) of these s-electrons.51 Therefore, when we are doping an alkali metal into the noble metal cluster, a systematic investigation of the excitations in molecular level is quite complicated. To this end, for investigating the absorption spectrum of the clusters we have presented the partial density of states (PDOS) calculation in Fig. 4, where the PDOS of Ag7, Ag5Li2, Ag2Li5 and Li7 clusters shown for clarification of the clusters optical spectra.
 | ||
| Fig. 4 Partial density of states of (a) Ag7, (b) Ag5Li2, (c) Ag2Li5 and (d) Li7 clusters. | ||
From the Fig. 4(a), it is evident that d electrons are more influential in Ag7 cluster with its peak range from −2 to −6 eV. On the other hand, this band has got narrower as a result of doping Li into Ag. For instance, in Ag5Li2 the d-electrons band got thinner ranged from −3 to −6 eV. It gets even narrower with the addition of more Li atoms, as we see the band comprises from −4 to −5 eV only in the case of Ag2Li5 clusters. Hence, it is obvious that the d-electrons are more dominant in the silver clusters but with the addition of alkali atoms, its dominance gets weaker. On the contrary, the s–p hybridization orbitals become stronger with the addition of Li and the influence of s–p hybridization is proportional to the number of Li atoms added into the system.
6. Conclusion
In this paper, the X (Li, Na) doped Ag nanoparticles are explored as potential improvement over Ag nanoparticles for plasmonic applications. For this work, X atoms are used to substitute Ag atoms from decahedral Ag7 structures to form AgmX7−m bimetallic clusters. Later, the ground state structures, chemical and optical properties of all the doped clusters are calculated in the framework of density functional theory. One of the targeted applications for this work is catalysis and it requires the materials to be of large band gap with very low electron affinity so that their anions become very reactive. Our calculation has found that the Li and Na doped clusters; especially the Ag4Li3 and Ag3Li4 clusters own high HOMO–LUMO gaps with low EA; hence these bimetallic clusters will be an excellent potential for catalytic applications. On the contrary, the calculated absorption spectrums of the AgmX7−m clusters have revealed that the doping of Li and Na has made the absorption band wider with regards to undoped Ag7 clusters. Typically opto-electronic devices, such as solar cells absorb lights in the range of 1–5 eV. With the calculations provided above, this work is suggesting that Li and Na doping (especially, Ag4X3, Ag3X4 and Ag2X5 clusters) will result in improvement of absorption band in the mentioned range. Consequently, the wider band will allow the increase of photo-current as well as the overall optical absorption efficiency. Hence, these Li and Na doped bimetallic nanoparticles can significantly improve the overall efficiency of opto-electronic devices such as thin film solar cells.Acknowledgements
The authors wish to acknowledge the University of Malaya-Ministry of Higher Education Grant UM.C/625/1/HIR/MOHE/ENG/29, University of Malaya Research Grant (UMRG) RP023B-13AET, University of Malaya Research Grant (UMRG) RP014C-13AET, and Malaysian Ministry of Science and Technology's Science Fund (06/01/03/SF0831) for the financial support. Deepest thanks also go to Professor Rene Fournier, Department of Chemistry, York University, Canada for the technical discussions and acknowledgment also to Prof. Dr Nasrudin Bin Abd Rahim and Universiti Malaya Power Energy Dedicated Advanced Centre (UMPEDAC) for the constructive support during the course of this work. The authors wish to thank Dr S. Balamurugan for his assistance in improving the text.References
- E. E. Zhurkin, T. van Hoof and M. Hou, Nanoscale alloys and core–shell materials: model predictions of the nanostructure and mechanical properties, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 224102 CrossRef.
- R. Ferrando, J. Jellinek and R. L. Johnston, Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- J. A. Rodriguez and D. W. Goodman, The Nature of the Metal–Metal Bond in Bimetallic Surfaces, Science, 1992, 257, 897–903 CAS.
- J. Chen, D. Wang, J. Xi, L. Au, A. Siekkinen and A. Warsen, et al., Immuno Gold Nanocages with Tailored Optical Properties for Targeted Photothermal Destruction of Cancer Cells, Nano Lett., 2007, 7, 1318–1322 CrossRef CAS PubMed.
- P. A. Derosa, J. M. Seminario and P. B. Balbuena, Properties of Small Bimetallic Ni–Cu Clusters, J. Phys. Chem. A, 2001, 105, 7917–7925 CrossRef CAS.
- D. Yuan, X. Gong and R. Wu, Peculiar distribution of Pd on Au nanoclusters: first-principles studies, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 035441 CrossRef.
- D. W. Yuan, Y. Wang and Z. Zeng, J. Chem. Phys., 2005, 122, 122114310 CrossRef PubMed.
- A. M. Schwartzberg and J. Z. Zhang, Novel Optical Properties and Emerging Applications of Metal Nanostructures, J. Phys. Chem. C, 2008, 112, 10323–10337 CAS.
- E. Janssens, S. Neukermans, H. M. T. Nguyen, M. T. Nguyen and P. Lievens, Quenching of the Magnetic Moment of a Transition Metal Dopant in Silver Clusters, Phys. Rev. Lett., 2005, 94, 113401 CrossRef CAS.
- N. C. Barnard, S. G. R. Brown, F. Devred, J. W. Bakker, B. E. Nieuwenhuys and N. J. Adkins, A quantitative investigation of the structure of RANEY®-Ni catalyst material using both computer simulation and experimental measurements, J. Catal., 2011, 281, 300–308 CrossRef CAS.
- K. Kaizuka, H. Miyamura and S. Kobayashi, Remarkable Effect of Bimetallic Nanocluster Catalysts for Aerobic Oxidation of Alcohols: Combining Metals Changes the Activities and the Reaction Pathways to Aldehydes/Carboxylic Acids or Esters, J. Am. Chem. Soc., 2010, 132, 15096–15098 CrossRef CAS PubMed.
- S. Duan and R. Wang, Bimetallic nanostructures with magnetic and noble metals and their physicochemical applications, Prog. Nat. Sci., 2013, 23, 113–126 CrossRef.
- Z. Liu, H. Lee, Y. Xiong, C. Sun and X. Zhang, Far-field optical hyperlens magnifying sub-diffraction-limited objects, Science, 2007, 315, 1686 CrossRef CAS PubMed.
- N. Fang, H. Lee, C. Sun and X. Zhang, Sub-diffraction-limited optical imaging with a silver superlens, Science, 2005, 308, 534–537 CrossRef CAS PubMed.
- T. W. Ebbesen, H. Lezec, H. Ghaemi, T. Thio and P. Wolff, Extraordinary optical transmission through sub-wavelength hole arrays, Nature, 1998, 391, 667–669 CrossRef CAS.
- T. Oates and A. Mücklich, Evolution of plasmon resonances during plasma deposition of silver nanoparticles, Nanotechnology, 2005, 16, 2606 CrossRef CAS.
- H. Wei and H. Eilers, From silver nanoparticles to thin films: evolution of microstructure and electrical conduction on glass substrates, J. Phys. Chem. Solids, 2009, 70, 459–465 CrossRef CAS.
- P. R. West, S. Ishii, G. V. Naik, N. K. Emani, V. M. Shalaev and A. Boltasseva, Searching for better plasmonic materials, Laser Photonics Rev., 2010, 4, 795–808 CrossRef CAS.
- M. G. Blaber, M. D. Arnold and M. J. Ford, Search for the Ideal Plasmonic Nanoshell: The Effects of Surface Scattering and Alternatives to Gold and Silver, J. Phys. Chem. C, 2009, 113, 3041–3045 CAS.
- M. G. Blaber, M. D. Arnold and M. J. Ford, A review of the optical properties of alloys and intermetallics for plasmonics, J. Phys.: Condens. Matter, 2010, 22, 143201 CrossRef CAS PubMed.
- B. Pietrobon and V. Kitaev, Photochemical synthesis of monodisperse size-controlled silver decahedral nanoparticles and their remarkable optical properties, Chem. Mater., 2008, 20, 5186–5190 CrossRef CAS.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- V. Bonačić-Koutecky, V. Veyret and R. Mitrić, Ab initio study of the absorption spectra of Agn (n = 5–8) clusters, J. Chem. Phys., 2001, 115, 10450–10460 CrossRef.
- R. Poteau, J.-L. Heully and F. Spiegelmann, Structure, stability, and vibrational properties of small silver cluster, Z. Phys. D: At., Mol. Clusters, 1997, 40, 479–482 CrossRef CAS.
- S. Debnath and S. M. Said, Structural and electronic properties of Li doped decahedral Ag 12 Li 1 bimetallic nanoclusters, in Semiconductor Electronics (ICSE), 2014 IEEE International Conference on, 2014, pp. 278–281 Search PubMed.
- S. Debnath, S. M. Said, M. F. Roslan, M. F. M. Sabri and B. D. Long, A DFT study on an alkali atom doped decahedral silver nanocluster for potential application in opto-electronics and catalysis, RSC Adv., 2015, 5, 7665–7672 RSC.
- A. Dębski, M. Braga and W. Gąsior, Calorimetric measurements and first principles to study the (Ag–Li) liquid system, J. Chem. Thermodyn., 2015, 82, 53–57 CrossRef.
- J. S. Pilgrim and M. A. Duncan, Photoionization spectroscopy of AgLi, Chem. Phys. Lett., 1995, 232, 335–340 CrossRef CAS.
- A. Stangassinger, A. M. Knight and M. A. Duncan, Photoionization spectroscopy of NaAg, Chem. Phys. Lett., 1997, 266, 189–194 CrossRef CAS.
- C. S. Yeh, D. L. Robbins, J. S. Pilgrim and M. A. Duncan, Photoionizaton electronic spectroscopy of AgK, Chem. Phys. Lett., 1993, 206, 509–514 CrossRef CAS.
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, Hardness conserving semilocal pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- E. K. U. Gross and W. Kohn, Time-Dependent Density-Functional Theory, in Advances in Quantum Chemistry, ed. L. Per-Olov, Academic Press, 1990, vol. 21, pp. 255–291 Search PubMed.
- R. Fournier, Theoretical study of the structure of silver clusters, J. Chem. Phys., 2001, 115, 2165–2177 CrossRef CAS.
- C. Jackschath, I. Rabin and W. Schulze, Electron impact ionization of silver clusters Ag n, n ≦ 36, Z. Phys. D: At., Mol. Clusters, 1992, 22, 517–520 CrossRef CAS.
- M. L. Tiago, J. C. Idrobo, S. Öğüt, J. Jellinek and J. R. Chelikowsky, Electronic and optical excitations in Ag n clusters (n = 1–8): comparison of density-functional and many-body theories, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155419 CrossRef.
- K. Chandrakumar, T. K. Ghanty and S. K. Ghosh, Static dipole polarizability and binding energy of sodium clusters Na n (n = 1–10): a critical assessment of all-electron based post Hartree–Fock and density functional methods, J. Chem. Phys., 2004, 120, 6487–6494 CrossRef CAS PubMed.
- J. Flad, H. Stoll and H. Preuss, Calculation of equilibrium geometries and ionization energies of sodium clusters up to Na8, J. Chem. Phys., 1979, 71, 3042–3052 CrossRef CAS.
- R. Fournier, J. B. Y. Cheng and A. Wong, J. Chem. Phys., 2003, 119, 9444–9454 CrossRef CAS.
- A. Nogga, P. Navrátil, B. Barrett and J. Vary, Spectra and binding energy predictions of chiral interactions for Li 7, Phys. Rev. C: Nucl. Phys., 2006, 73, 064002 CrossRef.
- J.-I. Aihara, Reduced HOMO–LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons, J. Phys. Chem. A, 1999, 103, 7487–7495 CrossRef CAS.
- P. W. Fowler and D. E. Manolopoulos, Magic numbers and stable structures for fullerenes, fullerides and fullerenium ions, 1992 Search PubMed.
- L. M. Molina and B. Hammer, The activity of the tetrahedral Au20 cluster: charging and impurity effects, J. Catal., 2005, 233, 399–404 CrossRef CAS.
- G. Gardet, F. Rogemond and H. Chermette, Density functional theory study of some structural and energetic properties of small lithium clusters, J. Chem. Phys., 1996, 105, 9933–9947 CrossRef CAS.
- W. Ma and F. Chen, Optical and electronic properties of Cu doped Ag clusters, J. Alloys Compd., 2012, 541, 79–83 CrossRef CAS.
- M. Moseler, B. Huber, H. Häkkinen, U. Landman, G. Wrigge and M. A. Hoffmann, et al., Thermal effects in the photoelectron spectra of Na N-clusters (N = 4–19), Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 165413 CrossRef.
- H.-C. Weissker and C. Mottet, Optical properties of pure and core–shell noble-metal nanoclusters from TDDFT: the influence of the atomic structure, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 165443 CrossRef.
- M. Harb, F. Rabilloud and D. Simon, Structural, electronic, magnetic and optical properties of icosahedral silver–nickel nanoclusters, Phys. Chem. Chem. Phys., 2010, 12, 4246–4254 RSC.
- M. Harb, F. Rabilloud, D. Simon, A. Rydlo, S. Lecoultre, F. Conus, V. Rodrigues and C. Félix, J. Chem. Phys., 2008, 129194108 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |