
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainability of cellulose dissolution and regeneration in 1,5-diazabicyclo[4.3.0]non-5-enium acetate: a batch simulation of the IONCELL-F process†
A.
Parviainen
a,
R.
Wahlström
b,
U.
Liimatainen
a,
T.
Liitiä
b,
S.
Rovio
b,
J. K. J.
Helminen
a,
U.
Hyväkkö
a,
A. W. T.
King
a,
A.
Suurnäkki
b and
I.
Kilpeläinen
*a
aDepartment of Chemistry, University of Helsinki, A. I. Virtasen Aukio 1, 00014 Helsinki, Finland. E-mail: ilkka.kilpelainen@helsinki.fi
bVTT – Technical Research Centre of Finland, Tietotie 2, 02044 Espoo, Finland
First published on 30th July 2015
Abstract
The recyclability of 1,5-diazabicyclo[4.3.0]non-5-enium acetate ([DBNH][OAc]), as a direct dissolution solvent for cellulose, was evaluated during laboratory scale recycling trials. The main objective was to simulate the conditions of a spinning bath from a Lyocell-type air-gap spinning process, called the IONCELL-F process. The saline solution was then concentrated, recycled and reused as many times as possible before cellulose dissolution was no longer possible. The chemical compositions of the ionic liquid and pulp were recorded throughout the experiments. The results of the experiments showed that [DBNH][OAc] can be recycled from aqueous media with an average recovery rate of 95.6 wt% using basic laboratory equipment, without any further process intensification or optimisation. The recycling of the ionic liquid did not change the chemical composition or degree of polymerisation of the recovered pulp but the colour of the regenerated pulps gradually darkened as the recycling times increased. The ionic liquid was found to hydrolyse 6.0–13.6 mol% per cycle, under these conditions. The build-up of the hydrolysis product, 3-(aminopropyl)-2-pyrrolidonium acetate, killed the dissolution feature at between 30.6–45.6 wt% hydrolysis product. The enzymatic digestibility of the regenerated pulp samples was studied with both a monocomponent endoglucanase and a cellulase mixture. The amount of residual [DBNH][OAc] in the regenerated pulps was determined, by both NMR and capillary electrophoresis. Although hydrolysis of the ionic liquid occurs, this study clearly shows potential for industrial application, with appropriate process equipment and recycling conditions.
Introduction
Looking at the projections of global population growth and rapidly industrializing nations in Asia and South America, it is intuitive that the consumption of natural resources and man-made commodities will escalate correspondingly in upcoming years. The annual demand for textile fibres is predicted to be at 133.5 million tons in 2030 (with an annual growth rate of 3.1%).1 Although synthetic fibres will cover a part of the increasing demand they still lack some of the desirable properties that cellulose-based fibres have (e.g. water management, water absorbency, recyclability and sustainable sourcing).1,2 Woody biomass offers great potential for the production of paper, fine chemicals and various chemical commodities, in addition to fibrous biomaterials (e.g. textile fibres) and biofuels.3–6 The high crystallinity of cellulose requires refined solvent systems for its homogenisation. The need for efficient, economical and ecological solvents for homogeneous lignocellulose processing is growing as the demand for biodegradable and renewable materials increase.Ionic liquids (ILs), often quoted as being molten salts below a melting point of 100 °C, having low vapour pressure and some being highly efficient in lignocellulose dissolution, are widely acknowledged as a prime candidates for future industrial processes.3,7 These are excellent media for cellulose dissolution. However, ILs are often quite expensive and in some cases toxic,8 which means that they must be recovered and recycled to a high degree. Regardless of the cellulose dissolution capability, without recyclability the benefit of using ILs in processes where products are of low value might not be attractive enough for industrial adaptation. Previously we introduced two new recyclability concepts into biomass dissolving ILs, one based on phase-separation9 and one based on distillation of the IL.10,11 This last concept introduced structures based on acid–base conjugate (protic) ILs e.g. 1,1,3,3-tetramethylguanidinium acetate ([TMGH][OAc]) and [DBNH][OAc]. From a wider range of acid–base conjugates,11 [DBNH][OAc] has shown a great processing potential in air gap fibre spinning (IONCELL-F).2,12 In the basic air gap spinning process, the dissolved cellulose ‘dope’ is drawn from the spinneret nozzle through an air gap to a regeneration bath filled with water, where the cellulose regenerates and the direct-dissolution solvent is extracted into the aqueous phase. The solvent is recovered from the water bath by evaporation of the water. The recovery rate of the direct-dissolution solvent is significant for sustaining process and cost efficiency. In the N-methyl-morpholine-N-oxide (NMMO) Lyocell process the solvent recovery of NMMO is >99% which should be considered as a bench-mark for alternative Lyocell process development.13 Consequently, the recovery ratio and recyclability feature of [DBNH][OAc] was evaluated in laboratory scale trials using conditions similar to IONCELL-F process. In the standard Lenzing AG Lyocell process pulp is mixed with NMMO with a high water consistency. This is then concentrated down to close to the NMMO monohydrate (NMMO·H2O) composition using a FILMTRUDER®.14 This essentially allows for homogenization of the pulp, prior to dissolution at the NMMO·H2O composition. A filmtruder is similar to a thin-film or wiped-film evaporator, operating at reduced pressure, and allows for short residence time during evaporation. During this preliminary work the recycling scheme was simulated by: (1) dissolution of pulp, (2) regeneration into water at the approximate temperature of the spinning bath in the IONCELL-F process, (3) filtration of the pulp, (4) evaporation in a rotary evaporator to low water content. As such, the process could be cycled using the recovered IL for each new dissolution step. It should be noticed at this point that the simulation was performed using laboratory-scale equipment and is a quite rough representation of the industrial process, especially in cellulose regeneration and antisolvent evaporation steps.
One additional pathway for the production of renewable materials, chemicals or fuels is to hydrolyse plant cell wall polysaccharides to produce monosaccharides, which are transformed into the actual end products by microbial fermentations. Lignocellulosic biomass, as such, is however very recalcitrant towards hydrolysis and pre-treatment methods are needed to render the lignocellulose into a more accessible form for enzymes or other catalysts. Regeneration of cellulose from IL has been shown to greatly increase its enzymatic digestibility.15 The concept has been demonstrated in numerous reports for cellulosic model substrates, such as microcrystalline cellulose (MCC),15 and lignocellulosic substrates including e.g. rice16 and wheat straw,17 corn cobs,18 wood meal,19 perennial grass20 and many others. Imidazolium-based ILs have mainly been investigated in these trials but recently also bio-based structures, such as cholinium-21,22 and amino acid-based23 ILs. One aim in this work was to study how pulp digestibility is affected by treatment with [DBNH][OAc]. Several studies have been conducted on IL pre-treatment combined with recycling the IL. Lozano et al.24 demonstrated excellent enzymatic digestibility of MCC after regeneration from [Bmim]Cl including each of five IL recycling cycles, noting that the complete and thorough removal of residual IL from the regenerated cellulose plays a key part in obtaining high enzymatic digestibility. In a study by Qiu and Aita the pre-treatment efficiency with [Emim][OAc], as measured by enzymatic digestibility, was shown to decrease with each IL recycling cycle when sugar cane bagasse was used as substrate. The study concludes that the optimal pre-treatment conditions vary with the different ILs and biomass used. Therefore, adjustments to the pre-treatment temperature and residence times might be beneficial.25 In these studies and other similar studies the IL was recycled by simply distilling out the anti-solvent (water), which has been shown to accumulate e.g. lignin in the IL if lignin-containing feedstocks are used.26 However, Wu et al.27 showed that [Emim][OAc] can be reused up to ten times in the pre-treatment of various lignocellulosic feedstocks, without removing the dissolved lignin from the IL between the pre-treatment cycles. Other IL recycling methods include distillation of conjugate acid–base pair ILs10 and phase-separation in biphasic IL-aqueous systems, achieved by creating a concentrated salt solution of the aqueous part.9
In this study, it was examined how many times pulp can be dissolved into and regenerated from [DBNH][OAc], which was recycled after each regeneration cycle by removing the anti-solvent (water). The recycled IL was analysed for dissolved oligosaccharides and important parameters of the regenerated pulp from each regeneration cycle were determined: changes in molecular weight distribution and saccharide composition, entrapped IL in the regenerated pulp, colour characteristics and enzymatic digestibility. The colour characterization is especially important for any potential textile application, in the case when the cellulose dope is produced for textile fibre spinning.2,28
Materials, methods and experimental
The cellulosic pulp used in this study was Eucalyptus urograndis prehydrolysis kraft (PHK) pulp (Cuen intrinsic viscosity (ISO 5351-1): 468 mL g−1; Mn = 79.8 kg mol−1; Mw = 268.7 kg mol−1; Bahia Speciality Cellulose, Bahia, Brazil) was ground by means of a Wiley mill to produce a homogeneous cotton-like material. 1,5-Diazabicyclo[4.3.0]non-5-ene (DBN) was purchased from Fluorochem Ltd and was distilled before use. Glacial acetic acid was purchased from Sigma-Aldrich. The dissolution/regeneration was performed in a 250 mL Syrris Globe® glass-jacketed reactor. The drying of the IL was performed in a Büchi rotary evaporator (>10 mbar) and a custom built high vacuum rotary evaporator (1–3 mbar) to remove traces of water. 1H, 13C and COSY NMR spectra was collected using Varian 300 MHz Unity spectrometer and processed with MestreLab Research, MestReNova 9.0 software.Synthesis of the [DBNH][OAc], dissolution of cellulose and spinning-bath simulation
65.38 g (0.526 mol) of freshly distilled DBN was weighed into the reactor. The airspace was then replaced with argon gas followed by slow addition of 31.61 g (0.526 mol) of acetic acid. The temperature of the exothermic reaction was kept below 70 °C during the addition. After the last of the acetic acid was added the reactor was sealed and left to mix for 30 minutes at 100 °C. The IL was characterized using 1H NMR in DMSO-d6 and MeOH-d4 (see ESI†). 7 wt% of PHK pulp was added to the reactor and the mixture was homogenised by stirring with a propeller blade mixer. Dissolution was confirmed to be complete after ∼1 h by examination of the solution using optical microscopy. For the regeneration of the dissolved pulp the reactor temperature was set to 75 °C and after settling, 400 mL of 15 °C water was added to the reactor. The gel-like regenerated material was broken down manually and further homogenized using a standard electric kitchen blender (stick model). The solution was filtered through a sinter glass funnel (Pore size 3) and evaporated in a rotary evaporator (down to 10 mbar) at 60 °C. The solid material was refluxed for 2–3 hours in ethanol (EtOH), filtered and evaporated dry. The two IL fractions were combined and further dried in a high vacuum rotary evaporator for 2 h at 60 °C, 1–3 mbar. The IL was then characterized using 1H NMR as described above.Analysis of regeneration liquid
The regeneration ionic liquid (RIL) samples were then analysed for any dissolved cello- or xylo-oligomers using a capillary electrophoresis (CE) method with pre-column derivatization.29 Internal standard (galactose, 20 mg l−1) was added to the RILs and each sample was diluted with milli-Q water to a final IL content of 20 wt% [DBNH][OAc]. The samples were derivatized by 4-aminobenzoic acid ethyl ester (ABEE) and then subjected to CE analysis as previously described28 but with the following modifications to the published method: the volume of buffer quenching the derivatization reaction was reduced to 0.3 mL (previously 0.5 mL to 0.5 mL sample liquid), the CE analysis was performed in constant current mode at – 66 μA and 10% (v/v) MeOH was added to the background electrolyte.Chemical analysis of the pulp samples
The regenerated pulp samples were analysed for changes in the molecular weight (Mw) of cellulose, saccharide composition and their content of residual IL after the treatment. The Mw of the pulp samples was analyzed as previously described.30 Briefly, the samples were subjected to a solvent exchange treatment including three treatments with EtOH, three treatments with N,N-dimethylacetamide (DMAc) and finally dissolved in dry 8% (w/v) LiCl/DMAc solution. After dilution and filtration the samples were analysed on a gel-permeation chromatography (GPC) system with 0.8% (w/v) LiCl/DMAc as eluent and refractive index detection using pullulans in the Mw range of 5900–16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1 as Mw standards.
000000 g mol−1 as Mw standards.
The saccharide composition and content of residual IL of the regenerated pulp samples were determined by doing an acid hydrolysis of the samples and then analysing the saccharides and [DBNH][OAc] concentrations in the acid hydrolysate. The pulp samples were milled with a Retsch UltraCentrifugal Mill ZM 200 to a final particle size of <0.5 mm with a rotor speed of 14000 rpm. The milling was preceded by cooling the samples with liquid nitrogen, to avoid heat damage during milling. The dry matter content of the milled samples was determined as the average mass loss by heating three parallel samples at 105 °C overnight.
Acid hydrolysis was carried out in two steps; first the milled pulp sample (50 mg) was hydrolysed in 70% H2SO4 (0.5 mL) at 30 °C for 60 min, then diluted with milli-Q water to a final H2SO4 concentration of 4% and subjected to a secondary hydrolysis step at 121 °C for 50 min. After cooling the samples their exact volume was adjusted by adding milli-Q water to a final volume of 25 mL. The monosaccharides in the acid hydrolysates were analysed by High-Performance Anion Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD) as previously described.31 The amount of residual [DBNH][OAc] was determined from the acid hydrolysates by CE using a P/ACE MDQ capillary electrophoresis instrument (Beckman–Coulter, Fullerton, CA, USA) equipped with a photodiode array (PDA) UV/Vis detector. The background electrolyte used was 200 mM ammonium acetate with its pH adjusted to 8.3 with NaOH and the following CE analysis conditions: separation voltage +25 kV; separation temperature 25 °C, detection wavelength 205 nm in direct detection mode; injection 0.5 psi for 10 s; capillary length (Ldet\Ltot): 50/60 cm and capillary inner diameter 50 μm. The acid hydrolysates were diluted 1:1 with milli-Q water as the only sample pre-treatment prior to analysis. The amount of [DBNH][OAc] was quantified against a standard series of [DBNH][OAc] dissolved in water.
Pulp colour
The colour characteristics of IL-treated pulp samples were determined in the 200–800 nm wavelength region by UV-Vis reflectance spectroscopy using a Perkin-Elmer Lambda 900 UV/VIS/NIR spectrometer equipped with an integrating sphere. Sample pellets were pressed from the milled pulps using a Thermo Spectra-Tech Qwik Handi-Press with a 7 mm die set, originally designed for pressing KBr sample pellets for FTIR analysis. The sample pellets produced contained 100 mg of milled pulp, had a final diameter of 7 mm and were 4–5 mm thick, which was anticipated to be enough to completely stop all UV-Vis transmittance during the measurement.In addition to recording UV-Vis reflectance spectra, the formation/disappearance of chromophores was monitored in more detail by constructing difference spectra, in which the starting pulp spectrum was subtracted from the spectra of the other samples. The k/s spectra were calculated according to the Kubelka–Munk eqn (1).32 Unlike reflectance, the light absorption coefficient (k) is directly proportional to the chromophore concentration, and can thus give quantitative information of light-absorbing groups. The light scattering coefficient (s) is more related to the physical properties of the pellet surface, and it was assumed to be constant in all the pellet samples.
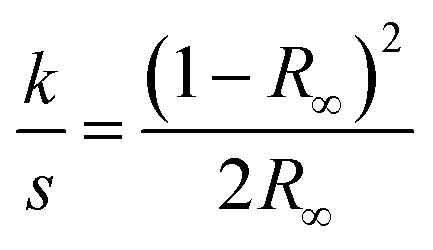 | (1) |
Enzymatic hydrolysis of the regenerated pulp samples
The non-milled regenerated pulps samples were subjected to enzymatic hydrolysis using two different cellulase preparations: (1) an enzyme cocktail for total hydrolysis was prepared by supplementing commercial Celluclast 1.5L (Novozymes) with β-glucosidase (Novozym 188, Novozymes) and (2) with a pure monocomponent preparation of Trichoderma reesei Cel5A (EG II). Cel5A was produced, isolated and purified at VTT as earlier described.33 The activities of the different cellulase preparations were determined according to previously described methods: Celluclast 1.5L total cellulose saccharification activity as filter paper units (FPUs),34 Novozym 188 β-glucosidase activity was determined on p-nitrophenyl-β-D-glucopyranoside35 and Cel5A endoglucanase activity on carboxymethylcellulose (CMC).30 The enzyme dosage in the total hydrolysis experiment was Celluclast 1.5L 2.5 FPUs per g substrate + Novozym 188100 nkatals per g substrate. The Cel5A dosage in the partial hydrolysis experiments was 2000 nkatals per g substrate. The hydrolyses were performed at 1% (w/v) substrate consistency at 45 °C under constant magnetic agitation, with a hydrolysis time of 24 h in 50 mM citrate buffer adjusted to pH 5.0 with NaOH. The dry regenerated pulp was swollen in buffer prior to hydrolysis. The hydrolysis was started by adding the cellulase and stopped by boiling the hydrolysis mixture for 10 min. After cooling to room temperature the hydrolysis mixtures were centrifuged and the clear supernatant was separated from the hydrolysis residue. The hydrolysis yield in terms of released reducing saccharides was determined from the supernatant by a 3,5-dinitrosalicylic acid (DNS) assay34 with the DNS reagent solution prepared as described by Sumner.36 The solid residues were washed and their Mw distribution analysed by GPC as described above.
Results and discussion
Spinning-bath simulation
In the Lyocell process, regeneration and media recycling conditions are roughly as follows: the dope temperature is 80–100 °C when it is regenerated into the spinning bath (10–30 °C) containing water. To recover the dissolving media bulk water is first removed to ∼20 wt% and then final water evaporation at 90–110 °C in a filmtruder. For this publication the process conditions were followed as closely as possible using a batch glass-jacketed reactor for regeneration. The media was held at 75 °C for the regeneration and was quickly cooled to room temperature to avoid excessive hydrolysis. Removal of water from the RIL was performed using rotary evaporators.Recycling experiments
Herein we shall introduce that the cation of [DBNH][OAc] is hydrolysed to some extent when in contact with water and at elevated temperatures. The mass recovery and the degree of hydrolysis (Fig. 1) were determined at each cycle. The total amount of hydrolysis per cycle was determined to be between 6.0 and 13.6 mol% using 1H NMR spectrometry. The degree of hydrolysis in the IL was 41.5–54.9 mol% when the dissolution capability was lost completely (between 5th and 6th cycle, 5th being the last to dissolve the sample completely). This corresponds to a water content of 4.1–5.4 wt% in the pure IL. The mass of the recovered IL was recorded during the recycling runs. The recovery of the IL by mass can be seen from Fig. 1. | ||
| Fig. 1 The mass of recovered IL and hydrolysis (wt%) after each cycle. | ||
Quantitative recovery of [DBNH][OAc]
For the process to be sustainable and efficient the recovery of the cellulose solvent needs to be high. The recoverability was evaluated in this simulation to get an initial prognosis on the use of [DBNH][OAc] as a processing solvent for cellulose. The recovery ratio of the process was calculated from the mass balances after recycling. The average recovery rate for the process solvent was 95.6%. (Note that the process solvent is a mixture of the initial [DBNH][OAc] and its corresponding hydrolysis product.) The process solvent recovery was the highest between 3rd and 7th cycles. Where the average recovery rate was 97.5% which corresponds to an average of 2.5 g mass loss for each cycle. These mass losses are attributed to working on such a small scale and with general laboratory equipment without further process optimisation. The actual recoveries should be close to 100%.Qualitative analysis of [DBNH][OAc]
In addition to recovery rate of the processing solvent it is necessary to investigate possible decomposition or side reactions during the recovery process. The NMR analysis showed that [DBNH][OAc] undergoes hydrolysis producing 3-(aminopropyl)-2-pyrrolidonium acetate ([APPH][OAc]), when it is heated with water as presented in Fig. 2. The NMR characterization of the IL and its corresponding hydrolysis product can be seen in Table 1.37,38 | ||
| Fig. 2 Hydrolysis of [DBNH][OAc] to [APPH][OAc]. | ||
| MeOH-d4 | DMSO-d6 | |||||||
|---|---|---|---|---|---|---|---|---|
| [DBNH][OAc] | [APPH][OAc] | [DBNH][OAc] | [APPH][OAc] | |||||
| 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C | |
| a The samples were run in MeOH-d4 relative to the solvent at 1H 3.31 ppm, 13C 49.0 ppm and in DMSO-d6 at 1H 2.50 ppm and 13C 39.52 ppm. | ||||||||
| 1 | — | — | — | — | — | — | — | — |
| 1′ | — | 180.0 | — | 180.1 | — | 173.8 | — | 174.1 |
| 2 | 3.46 | 43.5 | 3.39 | 40.5 | 3.30 | 41.9 | 3.20 | 39.1 |
| 2′ | 1.89 | 24.4 | 1.89 | 24.2 | 1.64 | 18.7 | 1.79 | 23.8 |
| 3 | 2.05 | 19.7 | 1.90 | 26.5 | 1.83 | 24.8 | 1.57 | 26.9 |
| 4 | 3.41 | 39.3 | 2.90 | 38.1 | 3.24 | 38.1 | 2.53 | 37.1 |
| 5 | — | — | — | — | — | — | 5.41 | — |
| 6 | — | 165.9 | — | 178.4 | — | 162.9 | — | 174.1 |
| 7 | 2.89 | 31.1 | 2.40 | 31.8 | 2.70 | 29.4 | 2.21 | 30.4 |
| 8 | 2.17 | 19.7 | 2.08 | 18.9 | 1.97 | 18.6 | 1.91 | 17.5 |
| 9 | 3.70 | 54.6 | 3.49 | 48.7 | 3.49 | 52.2 | 3.31 | 46.3 |
Because the C(7) proton signals from [DBNH][OAc] and [APPH][OAc] cations do not overlap with other signals, their integrals could be compared to estimate the state of the hydrolysis during the experiment (Fig. 3). The rate of the hydrolysis was determined to be roughly 6.0–13.6 mol% per cycle for the IL under these recycling conditions. A complimentary kinetic study of DBN and [DBNH][OAc] hydrolysis is still ongoing in our research group and will be published in the near future, but the preliminary results suggest that the hydrolysis does not promote an issue since it is reversible.
 | ||
| Fig. 3 Stacked 1H NMR spectra of the recycled [DBNH][OAc] showing the hydrolysis product build-up throughout the experiment. Solvent used was DMSO-d6. See ESI† for larger image. | ||
Determination of inorganic material build-up in the IL
Biomass contains residual inorganic material (e.g. salts) that originates from the soil, bound chemically and physically in the superstructure of the wood. In the typical Lyocell process, inorganic material bound to the initial pulp samples transfers into NMMO solution during dissolution and regeneration. The exact mass of inorganic material per cycle in this study was determined by adopting a microscale method published earlier by Hyväkkö et al.39 The result can be seen in (Fig. 4). | ||
| Fig. 4 Inorganic (ash) material build-up in the IL during recycling. | ||
Analysis of the regeneration liquid
In each pulp dissolution and regeneration cycle, the pulp was precipitated by adding water to the pulp solution in [DBNH][OAc]. After separating the precipitated pulp, the aqueous IL was dried by rotary evaporation and its content of dissolved oligosaccharides was analysed by a capillary electrophoresis method developed for saccharide analysis in aqueous IL solutions.29 This analysis was performed in order to study whether there was any significant degradation of the pulp polysaccharides to smaller soluble oligosaccharide fragments during the IL treatment. Such cellulose degradation has earlier been reported in combination with various IL treatments but the mechanism of this degradation process are still unclear.40,41 According to the CE analysis, no traces of soluble cello- or xylo-oligosaccharides were found in the RILs produced in this study, showing that the IL treatments with [DBNH][OAc] did not degrade the pulp polysaccharides to soluble oligosaccharides under the chosen regeneration conditions.Chemical analysis of the pulp samples
The saccharide composition of the pulp polysaccharides was determined after total acid hydrolysis of the untreated and IL-treated pulps (Table 2). The untreated pulp was almost pure cellulose as indicated by its high glucose content (97.7%) but it also contained some xylan (2.2%) and a very minor mannan fraction (0.1%). Surprisingly the saccharide composition stays very stable for the pulps treated in each cycle of pulp regeneration and is almost identical to the starting pulp. This result suggests that neither the cellulose nor the residual hemicelluloses in the used pulp are degraded or extracted by the regeneration treatment and all of the dissolved polysaccharide components appear to precipitate from the solution upon addition of the anti-solvent. This conclusion is also supported by the fact that no soluble saccharides could be detected in the RILs by CE. Previously published studies have described the extraction of hemicelluloses and lignin from lignocellulosic biomass as pre-treatment to enzymatic total hydrolysis of the plant cell wall polysaccharides.42–44| Glc | Xyl | Man | |
|---|---|---|---|
| Untreated pulp | 97.7 | 2.21 | 0.11 |
| 1 | 97.7 | 2.16 | 0.10 |
| 2 | 97.6 | 2.28 | 0.11 |
| 3 | 97.6 | 2.26 | 0.10 |
| 4 | 97.6 | 2.30 | 0.11 |
| 5 | 97.7 | 2.24 | 0.10 |
| 6 | 97.6 | 2.25 | 0.11 |
| 7 | 97.7 | 2.21 | 0.11 |
The entrapment of IL in regenerated cellulose during the regeneration procedure has been reported in several studies.45,46 Simply washing IL off the regenerated solid does not appear to remove IL entrapped in the regenerated matrix. IL entrapped in regenerated cellulose is a serious problem in different applications for various reasons: expensive IL is lost from the process leading to increased running costs, and many biomass-dissolving ILs are known to have moderate to high toxicities8 and should therefore not be present in significant amounts in end products, such as textiles. Finally, the presence of IL in IL pre-treated biomass is known to cause enzyme inactivation in enzymatic hydrolysis in biofuel and chemical production.46,47 The regeneration method used in the experiments does differ quite significantly from the projected fibre spinning process, based on the NMMO-based Lyocell process. From the perspective of removal of residual IL, the main difference is in the fact that, in the air-gap spinning process, the filament regenerating in the coagulation bath has a much larger surface area to volume ratio than the simulated method. Because of this it has to be noted that the residual solvent trapped during the process presented here, does not represent the state of the final fibres from a real large scale process.
For analysis, the pulp samples were subjected to total acid hydrolysis to degrade all the polysaccharides in the samples and release any trapped IL. The IL present in the acid hydrolysate was quantified by capillary electrophoresis. After IL-treatment, the solid pulp samples had been thoroughly washed with both water and EtOH meaning that any IL found in the hydrolysate originated from the matrix itself and not from liquid on the surface. The IL content of the IL-treated pulp samples varied greatly and did not appear to depend on the degree of pulp dissolution in [DBNH][OAc] (Table 3). High contents of IL were found in the regenerated pulps from cycles 1 and 2 (8.4% and 4.9% of the dry weight) whereas the pulps from cycles 3–5 contained 0.1–1.0% of IL. As could be expected, no IL was found from the untreated pulp or from the samples that were made after the point where dissolution capability was lost (samples 6 and 7). The IL amounts found in samples 1 and 2 are high and could pose a serious problem to the economics and fibre quality in the production of regenerated cellulose, if a method is not found to avoid the entrapment of IL in the cellulose matrix. The results obtained in this study do not allow speculating about the reason for the high IL content in pulps 1 and 2 compared to the other regenerated pulp samples. The residual IL determination was performed using quantitative 1H NMR with internal standard. The masses of residual IL in the regenerated pulp samples can be seen from Table 3.
| Sample | NMR | CE |
|---|---|---|
| Residual IL (m%) | Residual IL (m%) | |
| 1 | 5.6 | 8.4 |
| 2 | 2.3 | 4.9 |
| 3 | 1.1 | 0.7 |
| 4 | 0.3 | 0.1 |
| 5 | 0 | 1.0 |
| 6 | 0 | 0 |
The weight averaged Mw distribution of the untreated and IL-treated pulps was determined using GPC in LiCl/DMAc solution. The regeneration procedure did not change the Mw values of the pulps to any significant degree, taking into account the standard deviations between two parallel samples (Fig. 5). Obviously the pulps treated in the last two cycles, 6 and 7, which did not dissolve, also had similar Mw values as the other samples. The results demonstrate that the dissolution and regeneration method does not cause significant random cellulose hydrolysis under the applied conditions, which is an important prerequisite for e.g. fibre spinning processes in which cellulose Mw reductions should be avoided.
 | ||
| Fig. 5 Molecular weight values of untreated pulp (0) and pulps treated with recycled [DBNH][OAc] (1–7). Error bars indicate the standard deviation from between two parallel samples. | ||
Pulp colour
When producing regenerated cellulose in fibre spinning applications for textile use, the colour parameters of the produced fibres are important, why it was of interest to carefully monitor the development of colour in the regenerated material. The untreated pulp was a white and bright fibrous solid. It was, however, noticed by simple visual inspection, that a yellow or brown colour developed into the samples during the IL treatment (Fig. 6). The colour intensity gradually increased and darkened with the cycle number, the first three samples 1–3 clearly being less coloured than the samples from the later cycles. In addition, the texture of the samples was different. The untreated pulp was a fluffy cotton-like pulp, whereas the regenerated samples had a more compact form (sample 4 partly broke this trend). Samples 6 and 7, which did not dissolve, had a quite similar texture to the starting pulp but were coloured pale yellow by the IL treatment. To learn more about the nature of the colour origin, the samples were analysed by UV-Vis reflectance spectroscopy. In UV-Vis reflectance spectroscopy, the reflectance level correlates with the whiteness of the sample, so that a high reflectance level generally indicates a bright and white sample.48 The reflectance at the wavelength of 457 nm correlates well with the brightness values typically measured for market pulp or paper and it is very sensitive to the yellow colour caused by chromophores absorbing blue light. A comparison of the reflectance values at 457 nm (Table 4) strengthens the conclusions gained by visual inspection (Fig. 6.). Sample 4 had the lowest reflectance with 55% corresponding to the lowest brightness. The most significant finding was that the pulp regenerated in cycle 1 with fresh [DBNH][OAc] showed no reduction in brightness measured at 457 nm. The regenerated pulp samples 2–5 had approximately the same reflectance at 457 nm, whereas samples 6 and 7 had higher brightness, probably due to not having been dissolved. However, the IL treatment also caused some reductions to the brightness of the non-dissolved samples 6 and 7 as well, as compared to the untreated pulp. It should be noted that the colour could not be due to the IL itself, as the sample colour intensity and the amount of residual IL do not correlate but rather samples with high IL content had little colouration (Table 3). | ||
| Fig. 6 Appearance of non-milled untreated and in [DBNH][OAc] treated pulp samples. | ||
| Sample | Reflectance, % (457 nm) |
|---|---|
| Untreated pulp | 103 |
| 1 | 101 |
| 2 | 68 |
| 3 | 61 |
| 4 | 55 |
| 5 | 62 |
| 6 | 87 |
| 7 | 77 |
Theoretically, the chromophores causing the sample colour could be identified based on their absorption wavelengths, but often there are several absorbing structures in pulp and their quantities are very low, making their identification difficult. Rosenau et al.49–51 have made significant efforts in the identification of chromophores in technical pulps and those arising from the NMMO Lyocell process. The UV-Vis reflectance spectra show slowly rising curves with increasing wavelength (Fig. S1 see ESI†), not allowing identification of any single absorbing compound causing the colour, as this would be seen as a sharp peak in the spectral curve. The formation or disappearance of chromophores was monitored by constructing difference spectra, by subtracting the starting pulp spectrum from the other samples' spectra. In reflectance difference spectra, the structures formed into the starting pulp can be seen as positive signals and structures reacted/removed as negative signals. The difference reflectance spectra particularly show the changes occurring in the visible spectral region (400–800 nm). The IL-treated samples show the same reflectance difference shapes, although with varying difference reflectance levels, except sample 1, which appears to have lost some chromophore absorbing at 260–280 nm but gained none. The k/s spectra were calculated according to the Kubelka–Munk equation. Unlike reflectance, the light absorption coefficient (k) is directly proportional to the chromophore concentration, and thus yields quantitative information of light-absorbing groups. The absorption (k/s) difference spectra are especially sensitive to the changes in the UV region (200–400 nm). Although UV-active structures are colourless, they may function as precursors for coloured chromophores, and are therefore important to analyse. In difference absorbance spectra, the formed chromophores compared to the starting pulp can be seen as positive peaks and structures reacted/removed as negative peaks. By comparing the k/s spectra, formation of compounds absorbing UV-light at wavelengths 220 and 325 nm could be detected (Fig. S2 see ESI†). The peak at 325 nm in the k/s spectra clearly correlated with the sample colouration. Samples 1 and 2 had strong peaks at 220 nm whereas the more coloured samples had lower peaks in this region, strongly suggesting that the compound absorbing at 220 nm was not the compound responsible for sample colouration. In previous work, a correlation between signal intensity at 215 nm and pulp carbonyl content has been shown, and pulp carbonyl content is well known to contribute to the pulp brightness reversion. The exact origins of the absorptions at 220 and 325 nm are unknown but can be due to both degraded IL and degraded pulp components. A significant part of the DBN species in [DBNH][OAc] have been found to be hydrolysed to a structure containing an amide, which may explain the appearance of the 215 nm peak in the k/s spectra.
Enzymatic hydrolysis of the regenerated pulp samples
Lignocellulose regeneration from ILs has been shown to be an efficient means for increasing the enzymatic digestibility in plant cell wall polysaccharide hydrolysis. Two types of enzymatic hydrolysis experiments were done to assess the impact of the IL-treatment on the digestibility of the pulp samples. In total enzymatic hydrolysis experiments, a commercial cellulase cocktail, Celluclast 1.5L, supplemented with β-glucosidase, was used. In this setup, a rather low enzyme dosage (2.5 FPUs per g of substrate) was chosen to avoid enzyme over dosage and to thus have hydrolysis results more clearly describing the substrate digestibility properties. In an endoglucanase treatment with a purified monocomponent preparation of Trichoderma reesei Cel5A (EGII), the pre-treatment effect was assessed mainly as a measure of pulp crystallinity reduction. From the IL regenerated cellulose has been reported to have a high fraction of amorphous cellulose, which is the preferred substrate for endoglucanases.15 In recent studies, it has been proposed that cellulase cocktails should be reoptimized to contain significantly more endoglucanase activity for optimal performance on regenerated substrates.52,53 Endoglucanases are capable of causing rapid reductions in cellulose DP and especially if used on amorphous substrates, which is why the Mw distributions were followed in the hydrolysis experiments, in addition to yields of solubilised reducing saccharides.In the total hydrolysis experiments, the hydrolysis yields were moderately increased for the IL-treated pulps (Fig. 7). The pulp treated in cycle 2 had the highest digestibility (73%) whereas the untreated pulp had a digestibility of 45%, this is most probably because of the decrease in crystallinity due to the rough regeneration method used. Sample 4 did not show any increased digestibility although it had been dissolved during the IL treatment. The non-dissolved samples 6 and 7 showed approximately the same digestibility as the untreated pulp. In partial hydrolysis with T. reesei Cel5A the hydrolysis yields were much lower than in total hydrolysis as expected, but the differences between the yields from the different sample hydrolyses were relatively much greater than in the total hydrolysis, reflecting more clearly the difference in the pulp samples properties. The internal order of sample digestibility was the same as in the total hydrolysis, although sample 3 had a very low hydrolysis yield corresponding to that of sample 4 in partial hydrolysis.
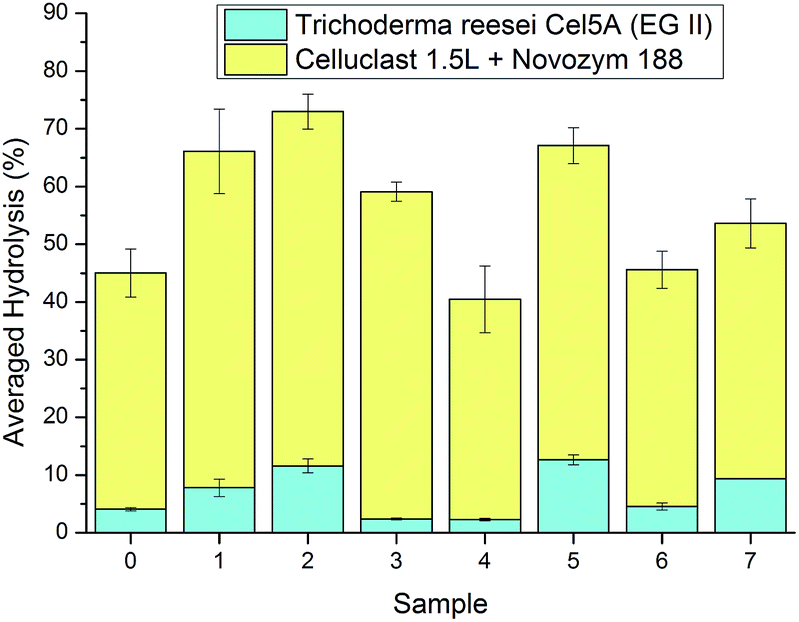 | ||
| Fig. 7 Yields from enzymatic hydrolysis of untreated and with [DBNH][OAc] treated pulp using Celluclast 1.5L supplemented with β-glucosidase (yellow) and Trichoderma reesei Cel5A (cyan). The hydrolysis temperature was 45 °C, the time 24 h and the hydrolysis medium 50 mM citrate buffer at pH 5.0. | ||
Samples 3 and 4 had low yields in the hydrolysis experiments possibly due to the change in crystallinity, which is avoided in a proper air-gap spinning process. The high contents of entrapped residual IL in samples 1 and 2 did not impede the enzymatic hydrolysis. In a previous study, [DBNH][OAc] was shown to be very toxic for T. reesei Cel5A at a concentration of 20 wt%, but in this work the amount of entrapped IL would amount to a maximum concentration of <1% if completely released to the hydrolysate and this low concentration is not likely to in any significant way affect the enzymes' performance.54 Sample colour and appearance did, however, to some extent correlate with digestibility. Sample 4 was clearly fluffier than 1, 2, 3 and 5, but had the lowest reflectance value at 457 nm, meaning it had the highest colour intensity. High colour intensity linked to a low hydrolysis yield could suggest enzyme inhibition by the substances causing pulp colouration or perhaps differences in the density of the pulp, which may also affect the colour. On the other hand, many other substrate parameters, which were not determined, are also likely to affect pulp digestibility. In terms of general enzymatic digestibility, it appears that the limiting factors are very sensitive to small variations in the regeneration conditions, which would explain the broken trend line in hydrolysis yields against regeneration cycle number. The working hypothesis was that the pulp dissolution and regeneration would be the most efficient in the first cycles and then fall when pulp components and degradation products accumulate into the recycled IL. This trend was only partly observed in this experiment, although the IL lost its ability to fully dissolve the pulp after the 5th cycle. It is clear that the properties of the pulp cannot be easily predicted for a specific regeneration process due to slight variation in experimental regeneration conditions. Studies in this area are needed and might yield interesting results for technical applications.
The influence of IL treatment on the enzymatic reduction of pulp DP was much greater than the difference observed in terms of hydrolysis yields. Generally, the regenerated pulps had higher DP reductions in the total hydrolysis treatments than in the partial hydrolysis endoglucanase treatments (Fig. 8). It is interesting to notice that pulps from cycles 6 and 7 had significant DP reductions even though they did not dissolve, suggesting non-dissolving IL treatments can still be effective pre-treatments for lignocellulose hydrolysis. For most of the samples, high hydrolysis yields were coupled with large DP reductions, but this was not observed for the undissolved samples 6 and 7. Though the DP reductions were greater in the total hydrolysis treatment than in the partial hydrolysis endoglucanase treatment, they generally followed the same trend, except for sample 3.
 | ||
| Fig. 8 Weight averaged molecular masses (Mw) of enzymatically hydrolysed untreated and [DBNH][OAc]-treated samples. Reference sample Mws are in yellow, total hydrolysis (Celluclast 1.5L supplemented with β-glucosidase) Mws in grey and partial hydrolysis (Trichoderma reesei Cel5A) Mws in cyan. The hydrolysis temperature was 45 °C, the time 24 h and the hydrolysis medium 50 mM citrate buffer at pH 5.0. | ||
Conclusions
During rough simulated dissolution and regeneration in a Lyocell process, it is clear that IL hydrolysis occurs and at some point prevents dissolution. This has been observed to occur at around 41.5–54.9 mol% of hydrolysis product, which corresponds to a 4.1–5.4 wt% of water impurity in the IL. However, if the IL recycling scheme can be optimised to avoid significant hydrolysis, the hydrolytic stability may actually be beneficial as it scavenges water from the reaction mixture that otherwise may have a more inhibitory effect on dissolution than the [APPH][OAc] itself. In the recycling scheme optimisation process it is clear that minimisation of the residence time of the aqueous IL on any hot evaporation surface is a must. The conditions reported here are far from optimum (rotary evaporator) as the residence times are very long. Therefore, thin-film evaporators, wiped-film evaporators and a filmtruder, rather than a batch distillation apparatus, should be used. Importantly, the continual dissolution, regeneration and IL recycling did not result in progressive pulp degradation, i.e. decrease in molecular weight and separation of hemicelluloses. This has been a limiting factor for other Lyocell processes and pulp degradation has been observed for other IL systems, in particular those based on the chloride anion.In terms of the chemical properties of the regenerated pulps, the increased hydrolysability of the regenerated cellulose most likely corresponds with an increase in amorphous cellulose or conversion to cellulose II. This is speculative as it was not directly confirmed but is common for cellulose dissolution and regeneration in direct-dissolution solvents. This can be seen as a positive feature for biomass pre-treatment purposes. The entrapped IL did not impede enzymatic hydrolysis of the regenerated pulp nor correlate with the darkening colour of the pulp, thus its main drawback was the loss of IL in the process. The somewhat uneven quality of the regenerated pulps shows that further studies are needed to learn how to design repeas and controllable pulp regeneration processes. In any case these batch regenerations are far from the actual air-gap spinning process, where the low fibre diameter at increasing draw ratios allows for much higher surface area to volume ratio to facilitate much more efficient removal of the IL.
Acknowledgements
The authors wish to acknowledge FiBiC (Finnish BioEconomy Cluster JR2 WP2) and TEKES (The Finnish Funding Agency for Technology and Innovation) for sponsorship.Notes and references
- F. M. Hämmerle, Lenzinger Ber., 2011, 89, 12–21 Search PubMed.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. K. J. Hauru, A. Parviainen, A. W. T. King, I. Kilpeläinen and H. Sixta, in Cellulose Chemistry and Properties: Materials, Fibers and Composites, ed. O. Rojas, Springer, Berlin, Heidelberg, 1st edn, 2015, Ionic Liquids for the Production of Man-Made Cellulosic Fibers: Opportunities and Challenges (online first) Search PubMed.
- I. Kilpeläinen, H. Xie, A. W. T. King, M. Granström, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- F. Wendler, B. Kosan, M. Krieg and F. Meister, Macromol. Symp., 2009, 280, 112–122 CrossRef CAS.
- F. Wendler, B. Kosan, M. Krieg and F. Meister, Lenzinger Ber., 2009, 87, 106–116 CAS.
- F. Wendler, L.-N. Todi and F. Meister, Thermochim. Acta, 2012, 528, 76–84 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- S.-K. Mikkola, A. Robciuc, J. Lokajová, A. Holding, M. Lämmerhofer, I. Kilpeläinen, J. Holopainen, A. W. T. King and S. Wiedmer, Environ. Sci. Technol., 2015, 49, 1870–1878 CrossRef CAS PubMed.
- A. Holding, M. Heikkilä, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2014, 7, 1422–1434 CrossRef CAS PubMed.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS PubMed.
- A. Parviainen, A. W. T. King, I. Mutikainen, M. Hummel, C. Selg, L. K. J. Hauru, H. Sixta and I. Kilpeläinen, ChemSusChem, 2013, 6, 2161–2169 CrossRef CAS PubMed.
- H. Sixta, A. Michud, L. K. J. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpeläinen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 43–57 CAS.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- S. Zikeli, B. Wolschner, D. Eichinger, R. Jurkivic and H. Firgo, European Patent, EP 1990 0356419 A2, 1990.
- A. Dadi, S. Varanasi and C. Schall, Biotechnol. Bioeng., 2006, 95, 904–910 CrossRef CAS PubMed.
- X. Hou, T. Smith, N. Li and M. Zong, Biotechnol. Bioeng., 2012, 109, 2484–2493 CrossRef CAS PubMed.
- Q. Li, Y. He, M. Xian, G. Jun, X. Xu, J. Yang and L. Li, Bioresour. Technol., 2009, 100, 3570–3575 CrossRef CAS PubMed.
- Q. Li, X. Jiang, Y. He, L. Yi, M. Xian and J. Yang, Appl. Microbiol. Biotechnol., 2010, 87, 117–126 CrossRef CAS PubMed.
- S. Lee, T. Doherty, R. Linhardt and J. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- P. Verdía, A. Brandt, J. P. Hallett, M. J. Ray and T. Welton, Green Chem., 2014, 16, 1617 RSC.
- X. Hou, T. Smith, N. Li and M. Zong, Biotechnol. Bioeng., 2012, 109, 2484–2493 CrossRef CAS PubMed.
- F. Chang, H. Wang, G. Chatel, G. Gurau and R. D. Rogers, Bioresour. Technol., 2014, 164, 394–401 CrossRef PubMed.
- K. Ohira, Y. Abe, M. Kawatsure, K. Suzuki, M. Mizuno and T. Itoh, ChemSusChem, 2012, 5, 388–391 CrossRef CAS PubMed.
- P. Lozano, B. Bernal, I. Recio and M. Belleville, Green Chem., 2012, 14, 2631–2637 RSC.
- Z. Qiu and G. Aita, Bioresour. Technol., 2013, 129, 532–537 CrossRef CAS PubMed.
- S. Lee, T. Doherty, R. Linhardt and J. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- H. Wu, M. Mora-Pale, J. Miao and T. V. Doherty, Biotechnol. Bioeng., 2011, 108, 2865–2875 CrossRef CAS PubMed.
- L. K. J. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471–4481 CrossRef CAS.
- R. Wahlström, S. Rovio and A. Suurnäkki, Carbohydr. Res., 2013, 373, 42–51 CrossRef PubMed.
- R. Wahlström, S. Rovio and A. Suurnäkki, RSC Adv., 2012, 2, 4472–4480 RSC.
- M. Tenkanen and M. Siika-aho, J. Biotechnol., 2000, 78, 149–161 CrossRef CAS PubMed.
- J. Smidt and C. Heitner, Advances in Lignocellulosic characterization, ed. D. S. Argyropoulos, TAPPI Press, Atlanta, 1999, pp. 179–199 Search PubMed.
- A. Suurnäkki, M. Tenkanen, M. Silka-aho, M. Niku-Paavola, L. Viikari and J. Buchert, Cellulose, 2000, 7, 189–209 CrossRef.
- T. Ghose, Pure Appl. Chem., 1987, 59, 257–268 CAS.
- M. Bailey and M. Linko, J. Biotechnol., 1990, 16, 57–66 CrossRef CAS.
- J. Sumner, J. Biol. Chem., 1924, 62, 287–290 CAS.
- F. Pereira, D. da Silva Agostini, R. Dias do Espírito Santo, E. deAzevedo, T. Bonagamba, A. Job and E. González, Green Chem., 2011, 13, 2146 RSC.
- R. Gonçalves, P. Abdelnur, V. Santos, R. Simas, M. Eberlin, A. Magalhães and E. González, Amino Acids, 2011, 40, 197–204 CrossRef PubMed.
- U. Hyväkkö, A. W. T. King and I. Kilpeläinen, BioResources, 2014, 9, 1565–1577 CrossRef.
- A. Nakamoto, Y. Shoda, M. Goto, W. Tokuhara, Y. Noritake, S. Katahira, N. Ishida, C. Ogino and N. Kamiwa, Bioresour. Technol., 2013, 135, 103–108 CrossRef PubMed.
- J. Bian, F. Peng, X. Peng, X. Xiao, P. Peng, F. Xu and R. Sun, Carbohydr. Polym., 2014, 100, 211–217 CrossRef CAS PubMed.
- K. Nimomiya, T. Yamauchi, M. Kobayashi, C. Ogino, N. Shimizu and K. Takahashi, Biochem. Eng. J., 2013, 75, 25–29 CrossRef.
- X. Hou, N. Li and M. Zong, Bioresour. Technol., 2013, 136, 469–474 CrossRef CAS PubMed.
- Q. Liu, X. Hou, N. Li, S. Ravula and H. Zhao, Green Chem., 2012, 14, 304–307 RSC.
- S. Xia, G. A. Baker, H. Li, S. Ravula and H. Zhao, RSC Adv., 2014, 5, 10586–10596 RSC.
- H. Zhao, C. L. Jones, G. A. Baker, S. Xia, O. Olubajo and V. N. Person, J. Biotechnol., 2009, 139, 47–54 CrossRef CAS PubMed.
- M. B. Tuerner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 443 RSC.
- M. A. Hubbe, J. J. Pawlak and A. A. Koukoulas, BioResources, 2008, 3, 627–665 Search PubMed.
- T. Rosenau, A. Potthast, P. Kosma, H. Suess and N. Nimmerfroh, Holzforschung, 2007, 61, 656–661 CAS.
- T. Rosenau, A. Potthast, W. Milacher, I. Adorjan, A. Hofinger and P. Kosma, Cellulose, 2005, 12, 197–208 CrossRef CAS.
- P. Korntner, T. Hosoya, T. Dietz, K. Eibinger, H. Reiter, M. Spitzbar, T. Röder, A. Borgards, W. Kreiner, A. Mahler, H. Winter, Y. Groiss, A. French, U. Hennings, A. Potthast and T. Rosenau, Cellulose, 2015, 22, 1053–1062 CrossRef CAS.
- P. Engel, S. Krull, B. Seiferheld and A. C. Spiess, Bioresour. Technol., 2012, 115, 27–34 CrossRef CAS PubMed.
- P. Engel, L. Hein and A. C. Spiess, Biotechnol. Biofuels, 2012, 5, 77 CrossRef CAS PubMed.
- R. Wahlström, A. W. T. King, A. Parviainen, K. Kruus and A. Suurnäkki, RSC Adv., 2013, 3, 20001–20009 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12386k |
| This journal is © The Royal Society of Chemistry 2015 |