
Direct templating assembly route for the preparation of highly-dispersed vanadia species encapsulated in mesoporous MCM-41 channel
Fu Yang,
Shuying Gao,
Cuirong Xiong,
Saifu Long,
Xiaoming Li,
Tao Xi and
Yan Kong*
State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 210009, China. E-mail: kongy36@njtech.edu.cn; Fax: +86-025-83587860; Tel: +86-025-83587860
First published on 3rd August 2015
Abstract
Understanding the nature of active sites, including the number and dispersion on the surface of a support, is essential to improve the catalytic activity. In this study, highly-dispersed and controllable quantities of vanadia species within the channels of mesoporous MCM-41 were directly prepared by a direct templating assembly method (S+L−M+I−). This method was based on the self-assembly of cationic surfactants (CTA+, S+), chelating agents (citrate ions, L−), vanadyl ions (VO2+, M+) and silicate oligomers (I−) via electrostatic and chelating interaction. First, the citrate ions were absorbed on the CTA+ micelles' surface by electrostatic interaction, and the vanadyl ions were subsequently anchored on their surface by chelating with citrate ions to form metallomicelles. Finally, the silicates were deposited on the metallomicelles to obtain the targeted product. The structures of the samples especially the oxidation state and surface distribution of vanadium species on the mesoporous silica were efficiently characterized with different techniques, including XRD, N2 adsorption, SEM, TEM, UV-vis, XPS, FT-IR, ICP, and H2-TPR. Furthermore, the samples obtained using hydroxylation of benzene as a probe reaction exhibited superior catalytic activities when compared with the post-synthesized sample.
1. Introduction
Mesoporous silica materials are well-known as a good carrier because of their oriented-ordered nanochannels and large surface areas1–7 and have been widely researched with functional modifications over the past two decades.8–14 A series of derived mesoporous composites have also shown potential applications in biomedical science,15 optical sensing,16 and catalysis.17–19Supported vanadium oxide catalysts contain the vanadium oxide phase deposited on mesoporous silica and have found extensive applications as oxidation catalysts in the chemical and petroleum industries. In particular, the supported vanadia phase, presented as surface vanadium oxide species, is responsible for the overall catalytic activity and selectivity.20 In addition, quantized catalytic active centers, such as particles or clusters, would be preferable to enhance the catalytic activity of supported vanadia in catalytic processes. Therefore, the dispersion and number of catalytically active sites on the supports would be the key factors that influence the catalytic activity of the catalysts. Many synthesis methods, including sorption, phase transitions, ion exchange, complexation and covalent grafting, have been utilized to modify mesoporous silica for the preparation of catalytic materials functionalized with metal species.21–26
These methods, however, require multistep synthesis processes and a large number of processes for modifying the raw material,27–30 resulting in destruction of the material's structure and regularity. Moreover, the precursors tend to be absorbed on the external surface of mesoporous silica, leading to large particles growing outside the mesopores.31 It was therefore highly desirable to seek a direct synthetic route for obtaining functional catalytic materials with high-dispersion and a high-loading of catalytically active sites.
Therefore, herein, we attempted to develop a simple and efficient templating synthetic method (S+L−M+I−) for the modification of micelles to directly obtain surface-functionalized mesoporous silica with highly-dispersed and controllable quantities of vanadia species. Through this method, we prepared metallomicelles (S+L−M+) with metal complexes attached on their surface by self-assembly of CTA+ (S+), citrate (L−) and VO2+ (M+) ions. We subsequently used them to synthesize our targeted mesoporous composites. In addition, different characterization methods were applied to test the structure of the materials and the oxidation state and distribution of vanadium oxides. Finally, benzene hydroxylation was employed as a probe reaction to evaluate the catalytic activities of the resultant samples.
2. Experimental section
2.1. Materials
Cetyltrimethylammonium bromide (CTAB, 99%), vanadyl sulfate (VOSO4, AR), vanadyl acetylacetonate (C10H14O5V, AR), tetraethyl orthosilicate (TEOS, AR), acetone (AR), sodium citrate (AR), ammonia solution (25 wt%), acetonitrile (AR) and benzene (AR) were obtained from Sinopharm Chemical Reagent Co., Ltd. Hydrogen peroxide (H2O2, 30 wt%) was purchased from Shanghai Chemical Corporation Pilot.2.2. Preparation of V/MCM mesoporous composites
The typical synthesis process involved four facile steps of mixing the surfactants, metallization of micelles, coating of silicates and calcination. An exemplary preparation process was as follows: 3.644 g CTAB (0.01 mol) was dissolved in 200 mL distilled water. The amount of vanadium in the composites was adjusted by controlling the molar ratio (VO2+/CTA+ = 0.2, 0.3, 0.4, 0.5, 0.6). Before adding VOSO4, an equimolar amount of sodium citrate was added to the CTAB solutions, and then different amounts of VOSO4 were added to the abovementioned mixed solutions with additional stirring for 30 min. The pH of the resulting solutions was adjusted to 10 with concentrated ammonia (25 wt%); subsequently, 11.15 mL of TEOS was rapidly added and stirred for 1 h under strong agitation. The obtained gel was then transferred to a Teflon stainless autoclave and aged at 100 °C for 3 days. The resulting samples were centrifuged and washed with distilled water and ethanol three times. Finally, the obtained dried samples were calcined at 550 °C for 5 h in a dry air stream at a heating rate of 1 °C min−1. The calcined samples were designated as xV/MCM (where x was 100 times the V/CTAB molar ratio).In contrast, another MCM-supported vanadia catalyst was prepared by a typical wet impregnation method. Briefly, pure MCM-41 carrier (0.15 g) was evenly dispersed in an ethanolic solution containing 0.074 g of vanadyl acetylacetonate (0.28 mmol) by ultrasonic treatment, and ethanol was rotary evaporated until complete dryness. The catalysts were then dried in air at 100 °C overnight and then calcined at 550 °C for 6 h in air to obtain the 60V/MCM sample (calculated molar ratio V/Si = 11.2%).
2.3. Characterization
The XRD patterns of the samples were collected with a Smartlab TM 9 KW (Rigaku Corporation, Tokyo, Japan) instrument equipped with a rotating anode and Cu Kα radiation (λ = 0.154178 nm).The N2 (77.4 K) adsorption–desorption measurements were carried out with a BELSORP-MINI volumetric adsorption analyzer (BEL Japan, Osaka, Japan) in the relative pressure (P/P0) range from 0.01 to 0.99. The annealed samples were outgassed under vacuum at 150 °C for 5 h and before measurements. The specific surface areas and pore size distributions were calculated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively.
High-resolution transmission electron microscopy (HRTEM) images were recorded on a JEM-2010 EX microscope (JEOL, Tokyo, Japan), which was operated at an accelerating voltage of 200 kV. The samples were crushed in A.R. grade ethanol and the resulting suspension was allowed to dry on a carbon film supported on copper grids.
Diffuse reflection UV-vis spectra of the samples were obtained in the range of 200–800 nm with a Lambda 950 spectrophotometer (Perkin Elmer, Waltham, MA, USA).
The X-ray photoelectron spectra (XPS) were acquired on a PHI 5000 Versa Probe X-ray photoelectron spectrometer (ULVAC-PHI, Kanagawa, Japan) equipped with Al Kα radiation (1486.6 eV). The C1s peak at 284.6 eV was used as the reference for the binding energy.
Fourier Transform Infrared (FT-IR) spectra of the samples were obtained in the range of 400–4000 cm−1 using powders dispersed in KBr and a Bruker VECTOR 22 resolution (Bruker, Germany) instrument.
The vanadium content in the samples was determined using a PE Optima 2000DV (Perkin Elmer, Waltham, MA, USA) Inductively Coupled Plasma (ICP) emission spectrometer. The samples were completely dissolved in hydrofluoric acid before analysis.
Hydrogen temperature programmed reduction (H2-TPR) measurements were performed utilizing a fixed-bed reactor under a flow of 10% H2/Ar gas mixture with a heating rate of 10 °C min−1 from room temperature to 750 °C. Before TPR analysis, the carbonate and hydrate impurities were removed by passing argon over the catalyst at a flow rate of 30 mL min−1 at 300 °C for 1 h, and the system was then cooled to room temperature. The amount of H2 uptake during the reduction was measured continuously using a thermal conductivity detector (TCD).
2.4. Catalysis performance tests
Assessment of the catalytic activity of V/MCM in the direct oxidation of benzene was performed as follows: 0.1 g catalyst, 2 mL benzene and 6.89 mL H2O2 were added into a 50 mL flask containing 15 mL acetonitrile. The mixed solution system was then stirred for 10 h at 30 °C in a water bath. The products from the reactions were finally analyzed with an SP-6890 gas chromatograph (Lunan Ruihong Chemical Instrument Co. Ltd, China) with a 0.32 mm × 30 m SE-54 capillary column. The conversion and selectivity of the products were calculated by the external standard method.To investigate the recycling properties of the directly synthesized and post-synthesized catalysts, the two solid catalysts were separated from the reaction mixture by centrifugation and used again in a fresh reaction.
3. Results and discussion
The electrostatic assembly approach has been proposed for synthesis of MCM-41.32 The primary pathway involves the direct co-condensation of anionic inorganic species (I−) with a cationic surfactant (S+), corresponding to assembled ion pairs from type S+I−. In this study, chelating agents (citrate ions) and metal cations (VO2+) were introduced to cooperatively assemble with the cationic surfactant and anionic inorganic species by electrostatic and chelating interactions.Scheme 1 illustrates the synthesis mechanism; the raw micelles consisted of CTA+, the tail of which carries electropositive ions and would spontaneously absorb the negatively charged citrate ions (L−). The added VO2+metal ions would then coordinate with citrate ions on the micelle surface to form the metallomicelles. Finally, deposition of silicate oligomers on the surface of the micelles would lead to formation of vanadium functional mesocomposites.
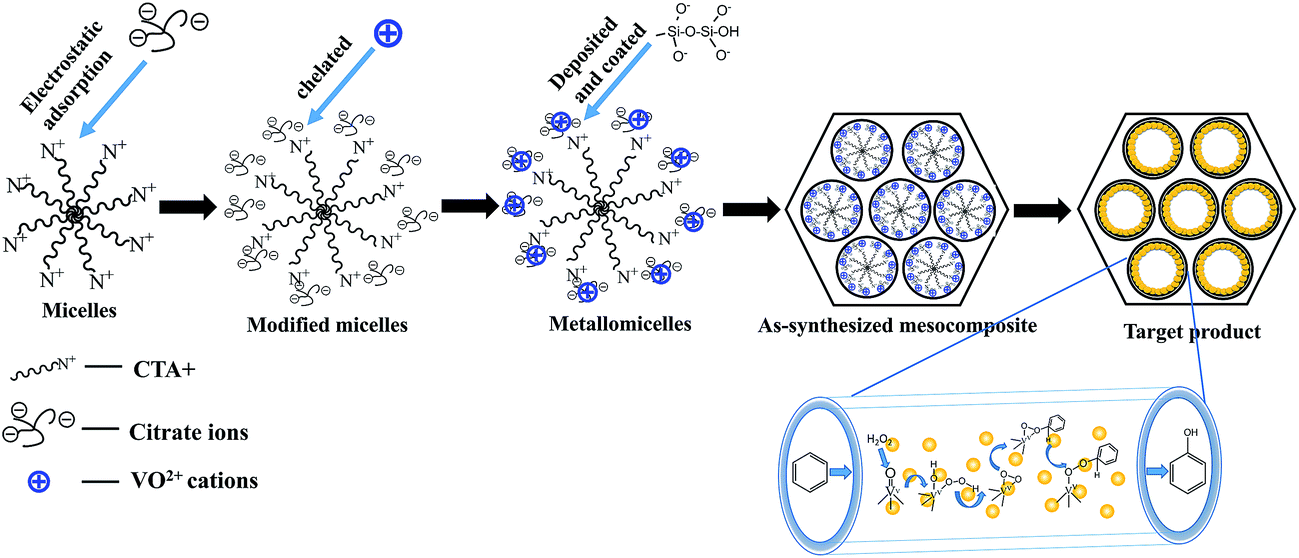 | ||
| Scheme 1 Mechanism for material self-assembly. | ||
3.1. Characterization of mesocomposite structure
The structural characteristics of all the samples were evaluated by low-angle XRD analysis (Fig. 1), N2 absorption/desorption measurements (Fig. 2) and SEM (Fig. 3). As shown in Fig. 1, all the prepared samples exhibited a strong (100) diffraction peak at 2θ = 2°–3°, indicating the mesoporous structure of the samples. In addition, the presence of two relatively weak 110 and 200 diffraction peaks at 2θ = 3°–5° suggested a highly ordered two-dimensional (2D) hexagonal structure with p6mm symmetry, which was typical for the MCM-41 mesoporous materials. These observations revealed that the highly ordered mesoporous structures of the host materials have been retained, even when vanadium oxides were encapsulated inside their matrices. Moreover, the three typical (100), (110) and (200) peaks showed a gradual shift to small angle with increasing vanadium contents in the samples, indicating an increase of the d100 interplanar spacing. This finding may be associated with enlargement of the metallomicelles by coordination with the increased number of vanadyl ions. | ||
| Fig. 1 Low-angle XRD patterns of samples containing different vanadium contents. | ||
 | ||
| Fig. 2 N2 adsorption/desorption isotherms (a) and pore size distribution (b) of samples containing different vanadium amounts. | ||
 | ||
| Fig. 3 SEM images of samples: (A) 20V/MCM, (B) 30V/MCM, (C) 40V/MCM, (D) 50V/MCM and (E) 60V/MCM. | ||
N2 adsorption/desorption isotherms (Fig. 2) showed that all the samples had typical type IV curves with an H1 type hysteresis loop, indicating that the synthesized samples were mesoporous materials with a two-dimensional hexagonal structure. In addition, more vanadium oxides were incorporated inside MCM-41 and the type IV shape of the isotherm was still maintained, suggesting retention of the ordered mesoporous structure. On the other hand, the spacing of the adsorption and desorption isotherms between relative pressures (P/P0) of 0.5 and 0.9 exhibited an increasing trend with increasing vanadium loading in the samples. This phenomenon could be attributed to increasing stacking pores between the particles due to the decreasing size of the particles with increasing vanadium loading, which is in accordance with SEM results. The more detailed structural characteristics of the samples are further summarized in Table 1. When we compared the pore size of different samples, we observed a gradual increasing trend in the pore diameter with increasing vanadium amounts. This result may be associated with the fact that increasing amounts of metal complexes were adsorbed on the surface of the micelles, leading to larger sizes of the metallomicelles. In addition, it can be noted that the pore wall thickness (dw) increased from 1.69 nm to 1.96 nm with increasing vanadium amounts in the samples. We propose that the increasing thickness could be assigned to a vanadium oxide layer loaded on the mesoporous silica MCM-41 pore walls.
| Samples | SBET (m2 g−1) | Pore volume (m3 g−1) | Pore sizea (nm) | Unit cell parameter, a0 (nm) (a0 = 2 × d100/√3) | d-spacing d100 (nm) | Pore wall thickness dW (nm) (dW = a0 − dP) |
|---|---|---|---|---|---|---|
| a The most probable pore size from pore size distribution calculated by BJH method using absorption data.b Sample represents synthesized pure MCM-41. | ||||||
| MCM-41b | 985 | 0.89 | 2.37 | 4.22 | 3.65 | 1.85 |
| 20V | 931 | 0.62 | 2.57 | 4.26 | 3.69 | 1.69 |
| 30V | 832 | 0.81 | 2.60 | 4.39 | 3.80 | 1.79 |
| 40V | 831 | 0.80 | 2.63 | 4.49 | 3.89 | 1.86 |
| 50V | 846 | 0.80 | 2.65 | 4.55 | 3.94 | 1.90 |
| 60V | 908 | 0.96 | 2.68 | 4.64 | 4.02 | 1.96 |
The morphology of the samples can be confirmed by the representative SEM images of the corresponding samples, and their results are shown in Fig. 3. Based on comparison of the samples containing different vanadium loadings, the size of the sample particles obviously became smaller with increasing vanadium loading. Accordingly, the number of aggregation voids became greater. This result supported the abovementioned N2 adsorption analysis. We propose that increasing loading of vanadium may influence the particles size of the samples.
In addition, more direct structural information could be obtained from the TEM images of the samples. The samples were evaluated using high-powered transmission electron microscopy and elemental mapping, and the results are shown in Fig. 4. The TEM results (Fig. 4a) demonstrated long-range order as well as a two-dimensional mesoporous structure with uniform channels, indicating that the metal oxides were embedded into the inner walls without damaging the regularity of the mesoporous phase. Moreover, the TEM images showed no evidence of large metal oxide particles, thus the vanadium oxides may be highly dispersed in the channel of mesoporous silica due to the confinement of the pore size. In addition, element mapping analysis was used to provide direct evidence of the presence and distribution of the vanadium oxide species. The element mapping images clearly show that the Si and O elements were uniformly and compactly distributed in the sample. The vanadium species were dispersed uniformly in mesoporous silica, which suggested high-dispersion of the vanadia species.
 | ||
| Fig. 4 TEM images (a) and elemental mapping (b) of 60V/MCM. | ||
3.2. Characterization of the state of vanadium oxide species
High-angle XRD patterns of all the samples are shown in Fig. 5. All the curves exhibited broad diffraction peaks from 20° to 30°, which can be ascribed to amorphous silica. Several other crystal diffraction peaks located at 37°, 43°, 64° and 77° could be detected by high-angle XRD, which can be assigned to vanadium oxide hydrate crystals (V2O5·xH2O). It can be noted that when the concentration of vanadium in the composites increased, the intensity of the diffraction peaks corresponding to vanadium oxide crystals gradually became stronger, indicating aggregation and increasing crystallinity of the vanadium oxide species in the directly synthesized mesocomposites. In addition, when compared with the directly synthesized samples, the diffraction peaks associated with the vanadium oxide crystals from the post-synthesized sample were obviously higher and sharper, indicating that much larger crystals were formed in the wet impregnation process. This fact suggested that the a smaller size of the quantized active species could be obtained due to the confinement of the narrow pore and interaction with the silicate walls for vanadium species in the direct synthesis method.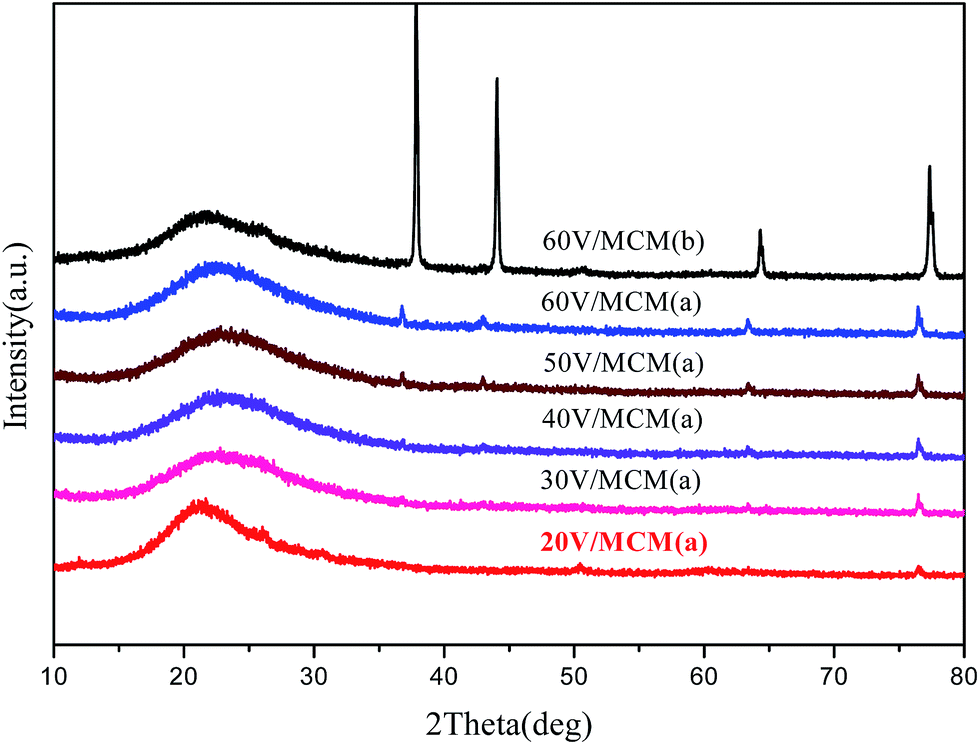 | ||
| Fig. 5 High-angle XRD patterns of samples containing different vanadium amounts: (a) directly synthesized sample and (b) post-synthesized sample. | ||
In addition, the state of vanadium in the materials was investigated using the diffuse reflection UV-vis spectra. Fig. 6 shows the diffuse reflection UV-vis spectra of the calcined V/MCM samples containing different vanadium amounts. All the samples showed two wide-ranges absorption peaks at 250 nm and 410 nm, indicating characteristic absorption of vanadium oxides. The presence of a broad band centered at ca. 250 nm was attributed to the surface isolated V5+ tetrahedral type VO43−, which was dispersed and interacted with the surface of the pore walls.33 This band is assigned to charge transfer (CT) associated with V–O electron transfer (π)t2 → (d)e and (π)t1 → (d)e. In addition, the featured peak at 410 nm shows the presence of clustered vanadium oxide species due to a further polymerization of the vanadium species,33 reflecting that vanadium oxide crystallites were formed in the materials.
 | ||
| Fig. 6 Diffuse reflection UV-vis spectra of samples containing different vanadium amounts. | ||
Further precise information on the oxidation states of surface vanadium species can be obtained from XPS data. Herein, X-ray photoelectron spectra of the vanadium 2p region and oxygen 1s region were studied for various samples with different vanadium loadings; the V2p and O1s core-level spectra are shown in Fig. 7a and b, respectively. Therein, the core level binding energy of the V2p3/2 and V2p1/2 peaks were located at ca. 517.0 and 524.6 eV, respectively. The binding energy at ca. 517.0 eV was assigned to V(5+) species in the sample, wherein the V2p3/2 peak was substantially enhanced and slightly shifted to a high binding energy.34 This result was associated with the fact that the VO43− species (corresponding to a relatively low binding energy) were first attached to the pore surface by bonding with silicon, after which increased clustering of V2O5 (corresponding to a relatively high binding energy) in the pore wall led to the shift to a high binding energy due to the decrease in the electron cloud density. In Fig. 7b, the main feature peak at ca. 533.0 eV was assigned to oxygen atoms bonded to silicon. Another small peak at ca. 529 eV assigned to oxygen atoms bonded to vanadium35 showed an increase in intensity with increasing vanadium content, indicating increasing numbers of vanadium oxide species on the pore walls.
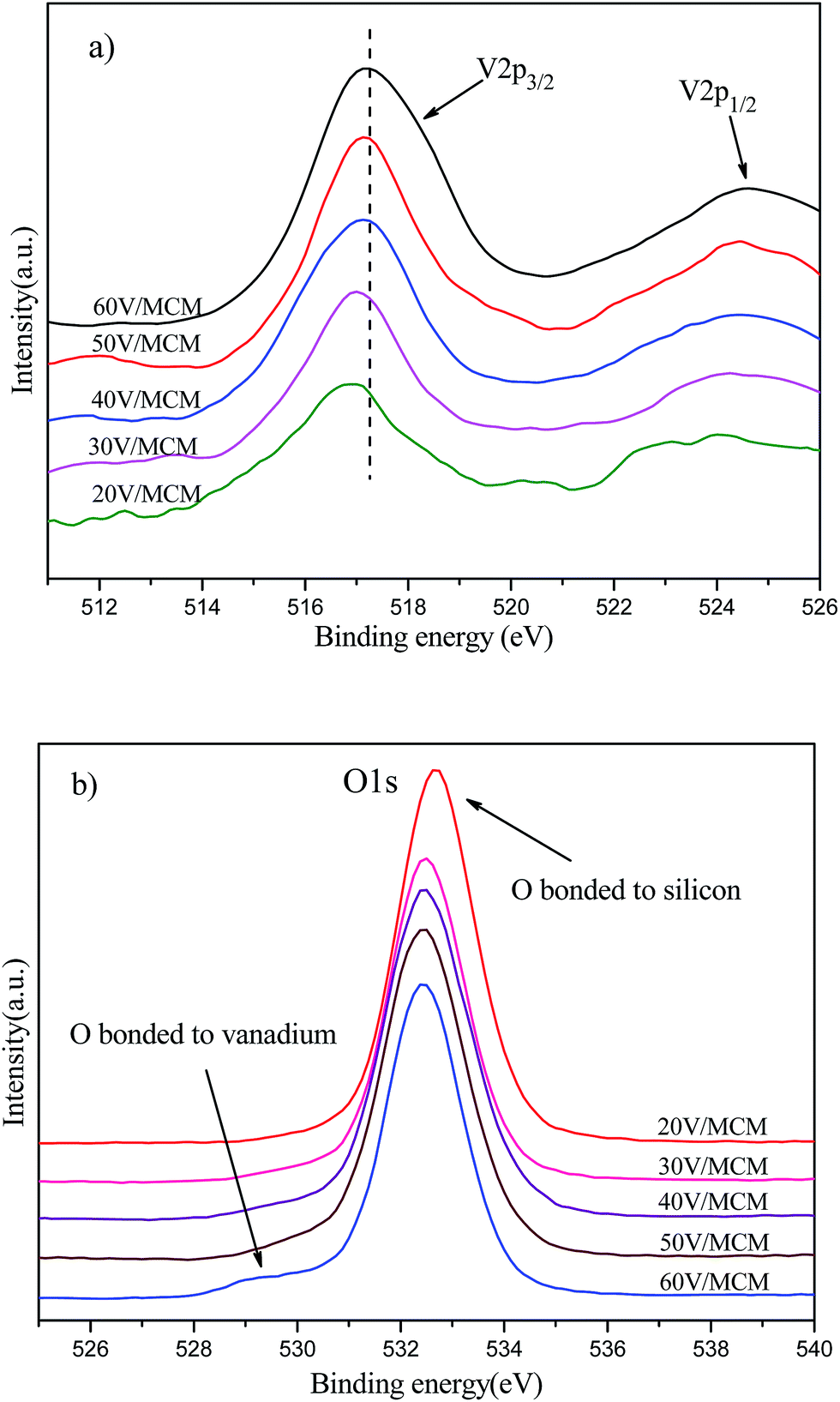 | ||
| Fig. 7 X-ray photoelectron spectra of samples with different vanadium amounts: (a) V2p3/2 (smoothing) and (b) O1s. | ||
The distribution of vanadium species on the pore walls can be reflected by the FT-IR spectra (Fig. 8). In general, the absorption peak at 966 cm−1 corresponding to the stretching vibration of Si–O–H surface groups36,37 became weaker with increasing vanadium concentration, implying a decreased amount of Si–O–H on the pore walls. The observation suggested that the surface Si–O–H groups were gradually covered with increasing vanadium oxide species. Moreover, the absence of surface Si–O–H may indicate that vanadium species interacted with the silicate walls. On the other hand, the FT-IR spectra of the 30V/MCM sample synthesized without citrate ions, the as-synthesized uncalcined 30V/MCM sample and sodium citrate are provided to prove the presence of citric acid. By comparison of the three aforementioned samples, sample B exhibited several additional absorption peaks at 3286, 1393 and 856 cm−1, which were in accordance with citrate. In addition, additional absorption peaks presented by the 30V/MCM sample exhibited a slight shift compared with pure citrate, which can indicate the existence of interaction in the sample.
 | ||
| Fig. 8 FT-IR spectra of samples with different vanadium amounts: (A) the 30V/MCM sample synthesized without citrate ions, (B) the as-synthesized 30V/MCM sample and (C) pure sodium citrate. | ||
The reduction temperature affects the size of the particle during the reduction process. Smaller particles are expected to be reduced at lower temperature, and a larger metal oxide particle size corresponds to a higher reduction temperature.27 Therefore, the H2-TPR profiles of the samples can give more information on the state of vanadium species on the pore walls of mesoporous silica. The H2-TPR patterns of the directly- and post-synthesized 60V/MCM samples are shown in Fig. 9. The results of temperature-programmed reduction are in agreement with high-angle XRD results (Fig. 1). The directly synthesized sample exhibited a sharp peak with maximum reduction temperature at Tm ca. 436 °C, whereas the post-synthesized sample showed a wider and relatively low peak with a maximum reduction temperature at Tm ca. 530 °C. The reduction temperature of the directly synthesized sample was significantly lower than that of the post-synthesized sample. This result can be ascribed to the fact that the fine and uniform vanadium oxide species were highly dispersed on the pore walls of mesoporous silica due to the confinement of the narrow pore. As for the post-synthesized sample, the high reduction temperature suggested that large metal oxide clusters may be formed on the external surface of the mesoporous MCM-41.33
 | ||
| Fig. 9 H2-TPR pattern of 60V/MCM samples (a) directly-synthesized (b) post-synthesized. | ||
3.3. Catalysis tests
In order to evaluate the catalytic properties of the series of V/MCM samples containing different vanadium contents, the direct oxidation of benzene was selected as a probe reaction (Scheme 2). The corresponding catalytic results are summarized in Table 2. The test results showed increasing conversion of benzene with increasing vanadium content in the samples. This result was apparently associated with the development of more accessible active sites on the surface of the catalyst due to increasing coverage of vanadium oxide species on the MCM-41 pore walls. After the V/Si molar ratio in the material reached 10.06%, the conversion of benzene did not improve significantly. We propose that the surface coverage of vanadium oxides on the pore walls became nearly saturated, which was in agreement with the FT-IR results. On the other hand, the directly synthesized 60V/MCM sample exhibited higher catalytic activities compared with that of the corresponding post-synthesized samples, 60V/MCM (P) and 60V/MCM (N). However, when the maximum vanadium amount was added into the post synthesis process for sample 60V/MCM (N), a corresponding high-loading of vanadium was not obtained for the sample synthesized without citrate. Moreover, compared with the directly synthesized sample 40V/MCM, we found that the sample 60V/MCM (N) did not provide higher conversion of benzene, even with a higher vanadium loading. This result may be ascribed to the fact that the metal atoms were incorporated in the framework of mesoporous silica, and the exposed active sites should be fewer than that in the directly synthesized sample. Finally, pure V2O5 crystals were used as a catalyst to catalyze the oxidation of benzene, and these crystals exhibited relatively low activities. This result indicated that the bulk V2O5 crystals possessed few catalytically active sites, whereas highly-dispersed vanadium (5+) species can efficiently catalyze the reaction. Herein, based on the abovementioned characterizations, we propose that highly-dispersed and high-loading catalytically active centers were produced in situ on the surface of the pore walls by the directed synthesis method, and the number of centers was significantly higher than that of the post-synthesized sample. Therefore, the catalytic results are also in favor of the proposed templating synthesis methods (Scheme 1).| Sample | V/Sib (%) | Benzene conversion (%) | Phenol selectivity (%) |
|---|---|---|---|
| a Reaction conditions: catalyst amount 0.1 g, benzene 2 mL, CH3CN 15 mL, 30% H2O2 6.89 mL, temperature 30 °C and duration 10 h.b V/Si molar ratios were analyzed by ICP.c Post-synthesized sample.d Sample was synthesized according to direct method without adding citrate. | |||
| 20V/MCM | 3.74 | 14.3 | 88 |
| 30V/MCM | 6.01 | 20.1 | 89 |
| 40V/MCM | 8.42 | 25.4 | 87 |
| 50V/MCM | 10.06 | 28.2 | 89 |
| 60V/MCM | 11.13 | 29.1 | 90 |
| 60V/MCM(P)c | 11.17 | 12.3 | 89 |
| 60V/MCM(N)d | 8.13 | 18.3 | 93 |
| V2O5 | — | 4.4 | 78 |
 | ||
| Scheme 2 Synthesis of phenol by direct oxide of benzene. | ||
In addition, the results of a recycling test using the directly- and post-synthesized samples for benzene hydroxylation are shown in Table 3. The post-synthesized sample did not exhibit reusability in the reaction, whereas the catalytic activity of the directly synthesized samples deteriorated slowly in comparison with that of the post-synthesized samples. We can thus conclude that the vanadium oxide species from the directly synthesized sample exhibited stronger interaction with the pore walls of the mesoporous silica, leading to more stable active centers in the catalyst. On the other hand, the results of N2 adsorption/desorption showed a gradual increase of the pore wall thickness for the series of directly synthesized samples, which may be another reason for the higher stability of catalyst because the high vanadium species surface coverage on the pore walls decreased the contact between the silicate walls and water in the reaction system.
| Samples | Runs | V/Sib (%) | Benzene conversion (%) | Phenol selectivity (%) |
|---|---|---|---|---|
| a Reaction conditions described as above, directly- and post-synthesized samples contain identical vanadium amount.b V/Si molar ratios were analyzed by ICP. | ||||
| Direct-60V/MCM | 1 | 11.13 | 29.1 | 90 |
| 2 | 8.13 | 23.2 | 89 | |
| 3 | 3.43 | 15.1 | 87 | |
| 4 | 1.45 | 8.3 | 89 | |
| Post-60V/MCM | 1 | 11.17 | 12.3 | 89 |
| 2 | 2.11 | 6.1 | 94 | |
| 3 | 0.12 | Trace | — | |
4. Conclusions
In summary, surface functional mesoporous silica encapsulating vanadium oxide species was prepared successfully via a direct templating assembly route. Surface functionalization of the silicate wall was closely associated with metallization of the micelles. The vanadium oxide species on the pore walls exhibited a high-dispersion and gradual coverage on the silicate surface with increasing vanadium concentration in the samples. The directly synthesized catalysts showed unique catalytic activities in comparison with the post-synthesized samples from benzene hydroxylation. This direct synthesis can potentially be utilized to prepare mesoporous materials containing other metal oxides and transition metal nanoparticles by modifying the micelle template. These composite materials have potentially extensive applications in catalysis.Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundations of China (21276125, 20876077 and 21476108).Notes and references
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS PubMed.
- K. Schumacher, C. D. von Hohenesche, K. K. Unger, R. Ulrich, A. Du Chesne, U. Wiesner and H. W. Spiess, Adv. Mater., 1999, 11, 1194–1198 CrossRef CAS.
- Y. Han, F. S. Xiao, S. Wu, Y. Y. Sun, X. J. Meng, D. S. Li, S. Lin, F. Deng and X. J. Ai, J. Phys. Chem. B, 2001, 105, 7963–7966 CrossRef CAS.
- B. Han, H. Wang, Y. Kong and J. Wang, Mater. Lett., 2013, 100, 159–162 CrossRef CAS PubMed.
- C. Wu, Y. Kong, F. Gao, Y. Wu, Y. Lu, J. Wang and L. Dong, Microporous Mesoporous Mater., 2008, 113, 163–170 CrossRef CAS PubMed.
- E. J. Acosta, C. S. Carr, E. E. Simanek and D. F. Shantz, Adv. Mater., 2004, 16, 985–989 CrossRef CAS PubMed.
- M. Alvaro, M. Benitez, D. Das, B. Ferrer and H. Garcia, Chem. Mater., 2004, 16, 2222–2228 CrossRef CAS.
- M. Alvaro, A. Corma, D. Das, V. Fornes and H. Garcia, Chem. Commun., 2004, 956–957 RSC.
- M. Alvaro, B. Ferrer, H. Garcia and F. Rey, Chem. Commun., 2002, 2012–2013 RSC.
- K. Ariga, Chem. Rec., 2004, 3, 297–307 CrossRef CAS PubMed.
- A. Liberman, N. Mendez, W. C. Trogler and A. C. Kummel, Surf. Sci. Rep., 2014, 69, 132–158 CrossRef CAS PubMed.
- P. Xu, X. Li, H. Yu and T. Xu, Sensors, 2014, 14, 19023–19056 CrossRef CAS PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- R. Metivier, I. Leray, B. Lebeau and B. Valeur, J. Mater. Chem., 2005, 15, 2965–2973 RSC.
- M. Mandal, V. Nagaraju, B. Sarma, G. V. Karunakar and K. K. Bania, ChemPlusChem, 2015, 80, 749–761 CrossRef CAS PubMed.
- R. Zubrzycki, J. D. Epping and T. Ressler, ChemCatChem, 2015, 7, 1112–1121 CrossRef CAS PubMed.
- H. Wang, W. Qian, J. Chen, Y. Wu, X. Xu, J. Wang and Y. Kong, RSC Adv., 2014, 4, 50832–50839 RSC.
- I. E. Wachs, Dalton Trans., 2013, 11762–11769 RSC.
- N. A. Melosh, P. Lipic, F. S. Bates, F. Wudl, G. D. Stucky, G. H. Fredrickson and B. F. Chmelka, Macromolecules, 1999, 32, 4332–4342 CrossRef CAS.
- H. Miyata, T. Noma, M. Watanabe and K. Kuroda, Chem. Mater., 2002, 14, 766–772 CrossRef CAS.
- B. J. Scott, G. Wirnsberger and G. D. Stucky, Chem. Mater., 2001, 13, 3140–3150 CrossRef CAS.
- M. Templin, A. Franck, A. DuChesne, H. Leist, Y. M. Zhang, R. Ulrich, V. Schadler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS.
- T. C. Xiao, S. F. Ji, H. T. Wang, K. S. Coleman and M. L. H. Green, J. Mol. Catal. A: Chem., 2001, 175, 111–123 CrossRef CAS.
- H. F. Yang, Q. H. Shi, X. Y. Liu, S. H. Xie, D. C. Jiang, F. Q. Zhang, C. Z. Yu, B. Tu and D. Y. Zhao, Chem. Commun., 2002, 2842–2843 RSC.
- C. Huo, J. Ouyang and H. Yang, Sci. Rep., 2014, 4, 3682 Search PubMed.
- K. Niu, L. Liang, H. Geng, W. Hou, H. Tian and S. Liu, Mater. Lett., 2013, 107, 325–328 CrossRef CAS PubMed.
- K. Niu, L. Liang, Y. Gu, L. Ke, F. Duan and M. Chen, Langmuir, 2011, 27, 13820–13827 CrossRef CAS PubMed.
- K. Niu, D. Shi, W. Dong, M. Chen and Z. Ni, J. Colloid Interface Sci., 2011, 362, 74–80 CrossRef CAS PubMed.
- H. F. Yang, Q. Y. Lu, F. Gao, Q. H. Shi, Y. Yan, F. Q. Zhang, S. H. Xie, B. Tu and D. Y. Zhao, Adv. Funct. Mater., 2005, 15, 1377–1384 CrossRef CAS PubMed.
- W. H. Zhang, M. Froba, J. L. Wang, P. T. Tanev, J. Wong and T. J. Pinnavaia, J. Am. Chem. Soc., 1996, 118, 9164–9171 CrossRef CAS.
- B. Solsona, T. Blasco, J. M. L. Nieto, M. L. Pena, F. Rey and A. Vidal-Moya, J. Catal., 2001, 203, 443–452 CrossRef CAS.
- H. Choi, J.-H. Bae, D. H. Kim, Y.-K. Park and J.-K. Jeon, Materials, 2013, 6, 1718–1729 CrossRef CAS PubMed.
- J. George, S. Shylesh and A. P. Singh, Appl. Catal., A, 2005, 290, 148–158 CrossRef CAS PubMed.
- E. Caponetti, A. Minoja, M. L. Saladino and A. Spinella, Microporous Mesoporous Mater., 2008, 113, 490–498 CrossRef CAS PubMed.
- B. L. Su, A. Leonard and Z. Y. Yuan, C. R. Chim., 2005, 8, 713–726 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |