
Controlled release of sunitinib in targeted cancer therapy: smart magnetically responsive hydrogels as restricted access materials
Abstract
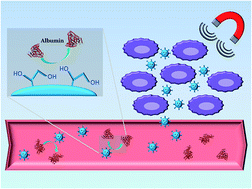
The aim of the present work was the preparation of a magnetic hydrogel for guided release of sunitinib malate (SUM), an anticancer drug. Precipitation polymerization method has been employed in order to synthesize the hydrogel, using methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA), glycidyl methacrylate (GMA) and 2,2′-azobisisobutyronitrile (AIBN) as monomer, cross-linker, pro-hydrophilic monomer and initiator, respectively. To confer magnetic responsiveness, particles of magnetite have been encapsulated during the polymerization process, followed by epoxide ring opening on the nanospheres' surface, in order to obtain a restricted access material (RAM) with low protein adsorption ability and improved biocompatibility. The successful introduction of magnetite has been confirmed through FT-IR and TG analyses, while protein adsorbing tests have been conducted to verify the RAM features. Furthermore, swelling properties have been evaluated before and after epoxide ring opening. Finally, in vitro tests have been performed to evaluate the release profile and cytotoxic effect on ARO, WRO, HeLa and MCF-7 cell lines.
 Please wait while we load your content...
Please wait while we load your content...

