
Interaction of 4-oxoalkane-1,1,2,2-tetracarbonitriles with Lawesson's reagent – a new approach to the synthesis of 2,2′-disulfanediylbis(1H-pyrroles). The synthesis of photochromic diarylethene with a disulfide bridge†
Abstract
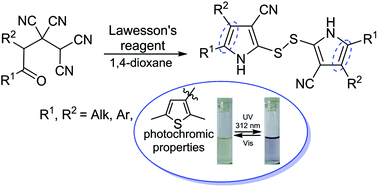
Transformation of 4-oxoalkane-1,1,2,2-tetracarbonitriles under the action of Lawesson's reagent leads to 2,2′-disulfanediylbis(1H-pyrrole-3-carbonitriles) in good yields. The developed method allowed the synthesis of photochromic 2,2′-disulfanediylbis(4,5-bis(2,5-dimethylthiophen-3-yl)-1H-pyrrole-3-carbonitrile).
 Please wait while we load your content...
Please wait while we load your content...

