
Functionalized multi-walled carbon nanotubes as an efficient reusable heterogeneous catalyst for green synthesis of N-substituted pyrroles in water†
Abstract
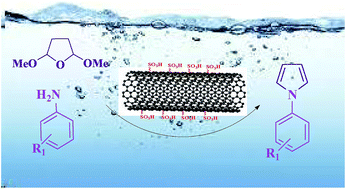
In this protocol, the functionalization of multi-walled carbon nanotubes (MWCNTs) was carried out and the resulting sulfonated MWCNTs were characterized by FT-IR, XRD, SEM, BET, EDX, XPS and Raman spectroscopy that are each discussed separately in the text. Then, the MWCNT–SO3H composite was applied as an efficient, recyclable heterogeneous catalyst for the synthesis of N-substituted pyrroles via the reaction of 2,5-dimethoxy tetrahydrofuran with primary amines under clean and mild conditions. In this reaction, the N-substituted pyrroles were obtained as beneficial and significant products in short reaction times (30–65 min) and good to excellent yields (40–92%) with high purity. The products were obtained through a simple work up procedure and characterized by FT-IR, 1H NMR and 13C NMR. After the end of the reaction, the nanocatalyst was recovered and reused several times without efficient loss of its activity for the preparation of N-substituted pyrroles.
 Please wait while we load your content...
Please wait while we load your content...

