
Stability study of tubular DNA origami in the presence of protein crystallisation buffer†
Abstract
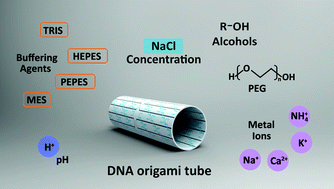
This work offers a methodology for screening compatible buffer conditions for both DNA origami and protein crystallisation and studied how protein crystallisation buffer conditions notably cations, buffering agents, precipitants, and pH, influenced the stability of tubular DNA origami.
 Please wait while we load your content...
Please wait while we load your content...

