
Triphenylamine-decorated BODIPY fluorescent probe for trace detection of picric acid†
Abstract
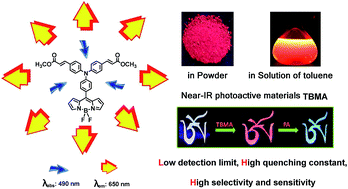
Triphenylamine-decorated BODIPY derivative TBMA was designed and synthesized. Luminogen aggregation was developed by taking advantage of twisted intramolecular charge transfer (TICT) and aggregation-induced emission (AIE) processes. In non-polar solvents, the locally excited (LE) states of BODIPY luminogens emitted intense yellow light. Increasing solvent polarity brought the luminogens from an LE to a TICT state, causing a large bathochromic shift in the emission color and a dramatic decrease in emission efficiency. The red emission was greatly boosted by aggregate formation or AIE effect. We also discovered that TBMA could be applied as an efficient chemical sensor for picric acid (PA) detection. The detection limit and quenching constant (KSV) were determined as 30 ppb and 2.1 × 10−6 M−1 respectively. 19F NMR and 1H NMR titration analysis verified that F⋯H hydrogen bonding is demonstrated as the mode of interaction, which possibly facilitates effective exciton migration.
 Please wait while we load your content...
Please wait while we load your content...

