
N-Urethane protection of amines and amino acids in an ionic liquid†
M. L. Di Gioia*,
A. Gagliardi,
A. Leggio,
V. Leotta,
E. Romio and
A. Liguori*
Dipartimento di Farmacia e Scienze della Salute e della Nutrizione, Edificio Polifunzionale, Università della Calabria, 87030 Arcavacata di Rende, Cosenza, Italy. E-mail: ml.digioia@unical.it; A.Liguori@unical.it; Fax: +39 0984 493265; Tel: +39 0984 493095, +39 0984 493205
First published on 20th July 2015
Abstract
An efficient, solvent-free protocol for the N-fluorenylmethoxycarbonylation and N-benzyloxycarbonylation of amines is described. The reaction of aliphatic and aromatic amines with FmocOSu and Cbz-Osu in [Bmim][BF4] at room temperature afforded the corresponding N-urethane derivatives in excellent yields and do not require any further purification. The method has been extended to the N-Fmoc and N-Cbz protection of amino acids. Absence of bases, very short reaction times, high yields, selectivity and ease of product separation are some advantages of this protocol.
Introduction
The development of mild and selective protocols for the protection and deprotection of amines continues to be an essential tool in the field of organic chemistry especially in peptide, nucleoside, polymer or combinatorial synthesis.1The presence of an amino group in various biologically active compounds makes its protection a necessity during their synthesis and transformation.1 Consequently, several kinds of protecting groups have been successfully used in this regard.2
In this context, the carbamate derivatives occupy a prominent position in the ranks of commonly used amine protecting groups (PGs).3
Among them, the use of carbamates, such as tert-butyloxycarbonyl (Boc), carbobenzyloxy (Cbz), and 9-fluorenylmethyloxycarbonyl (Fmoc), as protecting groups for amines has been significant because of the efficiency in the protection, inertness in various reaction conditions as well as easiness in deprotection.1,2a,3
The Fmoc group has been notably used for orthogonal protection of organic molecules for its important features of lability in basic media and stability in acidic media.4 The Cbz group is stable to basic and most aqueous acidic media and can be easily removed by catalytic hydrogenation.5 The Boc group enjoys popularity as a protecting group for amines due to its stability toward nucleophilic attack, basic media as well as catalytic hydrogenation.6
As a result of their importance, several methods for the preparation of Fmoc-7 Cbz-8 or Boc-9 protected amines have been developed to date. Nevertheless, all of these procedures have their own particular drawbacks, such as the use of expensive catalysts, strong basic conditions, long reaction times, the use of toxic (e.g. carcinogenic) solvents, chromatographic methods for the obtainment of the pure N-protected amines.
Hence, a simple, mild and environmentally friendly method for obtaining high yields in the urethane protection of amines remains highly desirable.
Room temperature ionic liquids (ILs), especially those based on the 1-N-alkyl-3-methylimidazolium cation, have shown great promise as smart alternatives to conventional solvents because of their unique features such as reasonable stability, low flammability, no miscibility with non-polar solvents.10
Due to their great potential as alternative reaction media for catalytic processes, much attention has been focused on organic reactions promoted by ionic liquids.11
In addition, ionic liquids show selectivity and enhancement in reaction rates, compared to common organic solvents with the added advantage of the ease of recovery and reuse of these ionic solvents.12 Due to these benefits, ionic liquids can make a substantial contribution to green chemistry.
Recently, we reported on the use of 1-butyl-3-methylimidazolium tetrafluoroborate [Bmim][BF4] as the ionic liquid medium for the clean removal of the 4-nitrobenzenesulfonyl (nosyl) protecting group and for the easy subsequent tertbutyloxycarbonylation of the free α-amino function of α-amino acid and dipeptide methyl esters.13 Moreover, various ILs catalysts have been employed for the preparation of N-Boc derivatives,14 and reports for the N-Cbz protection are scanty and require chromatographic purification;15 on the other hand there are no reports of using a ionic medium for the synthesis of Fmoc-protected amines in the literature.
In continuation of our efforts in green chemistry and to explore the applicability of ionic liquids as reaction media, here we report the successful use of [Bmim][BF4] as a solvent for the selective Fmoc and Cbz-protection of various structurally amines in a ionic liquid assisted process.
Results and discussion
In order to investigate the optimum model reaction, we selected, aniline (2a), an aromatic amine, as model substrate because of the low nucleophilicity of the amine nitrogen. aniline (2a) was treated with 9-fluorenylmethyloxycarbonyl chloride (Fmoc-Cl) (1) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mmol) in 1 mL of [Bmim][BF4]. The reaction was attempted at room temperature and in absence of any organic solvent or bases. TLC analysis of the reaction mixture showed after only 1 minute the disappearance of the FmocCl and the formation of a new spot on the TLC plate that travels with the solvent front and assigned to dibenzofulvene: in this case, it is more likely that the ionic liquid assists the β-elimination to give quickly the dibenzofulvene and liberation of the chlorine ion. Although the Fmoc-Cl (1) represents the reagent of choice traditionally employed for the incorporation of the Fmoc group, it has a series of drawbacks such as its instability and the tendency to promote the formation of undesirable by-products.16 Therefore, we decided to use a commercially available reagent with a different leaving group such as the 9-fluorenylmethyl succinimidyl carbonate (Fmoc-OSu) that has shown to be a more shelf-stable carbonate.17 We repeated the same model reaction using 1 equiv. of FmocOsu to add the Fmoc protecting group (Scheme 1). The reaction conducted in [Bmim][BF4] (1 mL) at room temperature was completed in 5 minutes and the expected N-(9-fluorenylmethoxycarbonyl)aniline (3a) was obtained in 86% yield after a simple extraction procedure with diethyl ether from the ionic liquid and without the need for chromatographic purification. The use of larger amounts of the IL did not improve the reaction yield or time.
:1 mmol) in 1 mL of [Bmim][BF4]. The reaction was attempted at room temperature and in absence of any organic solvent or bases. TLC analysis of the reaction mixture showed after only 1 minute the disappearance of the FmocCl and the formation of a new spot on the TLC plate that travels with the solvent front and assigned to dibenzofulvene: in this case, it is more likely that the ionic liquid assists the β-elimination to give quickly the dibenzofulvene and liberation of the chlorine ion. Although the Fmoc-Cl (1) represents the reagent of choice traditionally employed for the incorporation of the Fmoc group, it has a series of drawbacks such as its instability and the tendency to promote the formation of undesirable by-products.16 Therefore, we decided to use a commercially available reagent with a different leaving group such as the 9-fluorenylmethyl succinimidyl carbonate (Fmoc-OSu) that has shown to be a more shelf-stable carbonate.17 We repeated the same model reaction using 1 equiv. of FmocOsu to add the Fmoc protecting group (Scheme 1). The reaction conducted in [Bmim][BF4] (1 mL) at room temperature was completed in 5 minutes and the expected N-(9-fluorenylmethoxycarbonyl)aniline (3a) was obtained in 86% yield after a simple extraction procedure with diethyl ether from the ionic liquid and without the need for chromatographic purification. The use of larger amounts of the IL did not improve the reaction yield or time.
 | ||
| Scheme 1 | ||
These preliminary results were very promising as they clearly showed evident advantages of the procedure with respect to those reported in the literature: the Fmoc protection of the amino group efficiently occurs in the ionic liquid without the addition of bases, under solvent-free conditions and at room temperature. Furthermore, the urethane protected amine is afforded in very short time of reaction, after a simple solvent extraction procedure from the IL and without the need for chromatography. So, the ionic liquid [Bmim][BF4] acts as the solvent and probably as the catalyst by the electrophilic activation of Fmoc-OSu, making the carbonyl group susceptible to nucleophilic attack by the amine.
At this point, we attempted to extend the generality of the reaction by using [Bmim][BF4] as the reaction medium. Different aliphatic (acyclic and cyclic) amines and various aromatic and heteroaromatic amines were tested. The reaction thus carried out in [Bmim][BF4] at room temperature afforded the corresponding N-Fmoc derivatives in excellent yields and with a short reaction time (3–9 min) (Table 1).
|
||||
|---|---|---|---|---|
| Entry | Amine (2) | Product (3 and 4) | Time (min) | Yield (%) |
| a Reaction conditions: amine (1.0 mmol), FmocOSu (1.0 mmol), [Bmim][BF4] (1 mL), 25 °C.b Isolated yields.c 0.5 mL of saturated NaHCO3 solution was used as an additive.d Yield of the first of five runs using the same recovered IL. Yields of the four subsequent runs were: 91, 89, 88, 86, respectively. | ||||
| 1 |  |
 |
5 | 86 |
| 5 | 87 | |||
| 2 |  |
 |
8 | 85 |
| 8 | 88 | |||
| 3 |  |
 |
3 | 88 |
| 5 | 88 | |||
| 4 |  |
 |
7 | 88 |
| 7 | 89 | |||
| 5 |  |
 |
6 | 87 |
| 7 | 86 | |||
| 6 |  |
 |
6 | 89 |
| 6 | 86 | |||
| 7 |  |
 |
5 | 85 |
| 6 | 87 | |||
| 8 |  |
 |
3 | 91d |
| 3 | 95 | |||
| 9 |  |
 |
4 | 93 |
| 4 | 92 | |||
| 10 |  |
 |
3 | 96 |
| 3 | 95 | |||
| 11 |  |
 |
3 | 97 |
| 3 | 95 | |||
| 12 |  |
 |
5 | 89 |
| 5 | 87 | |||
| 13 |  |
 |
4 | 87 |
| 4 | 91 | |||
| 14 |  |
 |
9 | 88 |
| 8 | 86 | |||
| 15 |  |
 |
8 | 85 |
| 6 | 92 | |||
| 16 | 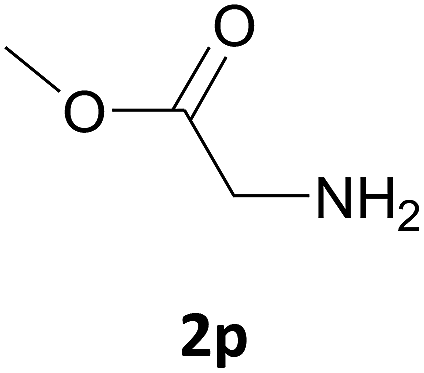 |
 |
9 | 90c |
| 9 | 87c | |||
| 17 |  |
 |
9 | 85c |
| 7 | 86c | |||
| 18 |  |
 |
8 | 86c |
| 8 | 90c | |||
| 19 |  |
 |
8 | 85c |
| 8 | 88c | |||

In most cases, the products were obtained in good to excellent yields. Aniline and benzylamine derivatives were subjected to the above described procedure affording the respective N-protected amines in good yields (Table 1, entries 1–5). In general, derivatives of benzylamine reacted faster than aniline derivatives because of their better nucleophilic character.
We also explored a wide variety of aliphatic acyclic and cyclic amines under the same standard reaction conditions. Notably, in these case the reactions proceeded very fast and in nearly quantitative yields (Table 1, entries 6 and 8–13). For instance, when piperidine was used the desired product was obtained in 97% yield and in only 3 minutes (Table 1, entry 11): secondary amines, in fact, generally took less reaction time due to better nucleophilicity. The existence of a double bond in the starting amine does not affect the protocol: when we used allylamine, the corresponding product was delivered in 85% yields in 8 minutes (Table 1, entry 15). From these results, it is evident that the studied IL, [Bmim][BF4], is a mild, highly efficient and a green catalyst for N-Fmoc protection of amines.
Excellent chemoselectivity was observed in case of an aminoalcohol as the starting material: in fact, in the case of ethanolamine and of tyramine (Table 1, entry 14, and 7 respectively) no bis Fmoc derivatives or mixture of O/N Fmoc-derivatives was observed. N-Fmoc-ethanolamine (3n) and N-Fmoc-tyramine (3g) were obtained in good yields (88 and 85%) and in fast reaction rate.
It is well known that masking the α-amino function of amino acids with urethane protecting groups has become topic of fundamental importance in the preparation of building blocks both in solution as well as solid phase peptide synthesis. So, after the effective results in the N-Fmoc protection of amines we have further extended our study by applying the protocol to the Fmoc protection of the α-amino group of amino acid methyl esters.
Nevertheless, our protocol was not applicable for the Fmoc protection of L-valine methyl ester hydrochloride (2r) due to the lack of solubility and nucleophilicity of the amino acid methyl ester as a HCl salt in the ionic medium. The best results were obtained after addition to the mixture of 0.5 mL of an aqueous solution of sodium bicarbonate therefore allowing the liberation of the amino group, which can then act as a good nucleophile for the protection reaction. Under the optimized reaction conditions, the Fmoc protection of valine methyl ester (2r) was thus achieved in 8 minutes and in 86% yield (Table 1, entry 18). Similarly, the treatment of other α-amino acid methyl esters with FmocOsu in [Bmim][BF4] using NaHCO3 as an additive provided the respective N-protected compounds in good to very good yields (Table 1, entries 16, 17 and 19).
Encouraged by these promising results, the developed protocol was also attempted to introduce the Cbz urethane group, by using N-(benzyloxycarbonyloxy)succinimide (Cbz-Osu) (5). Amines 2a–s were treated with 1 equiv. of CBz-Osu in [Bmim][BF4] under solvent-free conditions at room temperature and without the addition of bases (Scheme 2).
 | ||
| Scheme 2 | ||
Also in this case, most of the aliphatic and aromatic amines we checked gave high yields of carbamate (Table 1, products 4a–4o). In addition, the times required for derivatization were short and no chromatographic purification was necessary. Furthermore, the protocol showed to be highly chemoselective as the in the case of amino alcohols (Table 1, entries 4g–4n) the amine group is only protected even in the presence of OH groups and the N-Cbz protected compounds were obtained in excellent yields as the sole product without the competitive formation of the O-Cbz product. Again, esters of amino acids required 0.5 mL of a sodium bicarbonate aqueous solution as additive in the IL assisted protection process (Table 1, products 4p–4s).
The results of the presented protocol show that the ionic liquid [Bmim][BF4] is an efficient reaction medium as well as a catalyst for the N-Fmoc and N-Cbz protection of the amino group in amines and amino acid methyl esters.
The feasibility of any catalytic process is influenced by the possibility to reuse again and again the same catalyst. Therefore, the reusability of [Bmim][BF4] was assessed by conducting the N-Fmoc protection of cyclopentylamine (2h) over four successive runs. The results shown in Table 1 show no significant loss of activity of the IL.
Conclusions
In summary, we have reported a simple and convenient protocol based on the use of [Bmim][BF4] for the preparation of Fmoc and Cbz-protected amines and amino acid methyl esters. In particular, the absence of organic solvents or bases, the use of mild conditions, short reaction times, the high yields, the simple work-up procedure and finally the possibility to recycle the ionic liquid represent important features and advantages of the method providing a valuable contribution to the existing methodologies for the introduction of urethane amine protecting groups.Experimental section
General
Commercially available reagents were purchased from Sigma-Aldrich Chemical Co. (Milano, Italy) and used as supplied unless stated otherwise. Solvents were purified and dried by the standard procedures and distilled prior to use. All syntheses were carried out in atmospheric conditions. All the α-amino acids were of the L-series. 1H NMR spectra were recorded at 300 MHz, while 13C NMR spectra were measured at 75 MHz. Spectral analysis was performed at 293 K on diluted solutions of each compound by using CDCl3 as the solvent. Chemical shifts (δ) are reported in ppm and referenced to CDCl3 (7.25 ppm for 1H and 77.0 ppm for 13C spectra). Coupling constants (J) are reported in Hertz (Hz). Reaction mixtures were monitored by thin layer chromatography (TLC) using Merck Silica gel 60-F254 precoated glass plates, and UV light (254 nm) or 0.2% ninhydrin in ethanol and charring as visualizing agent. Evaporation of solvents was performed at reduced pressure using a rotary vacuum evaporator. LC-MS analysis was carried out using an Agilent UHPLC instrument coupled to a QTOF mass spectrometer fitted with an electrospray ionization source (ESI) operating in positive ion mode. GC-MS analyses were performed using a 30 m × 0.25 mm, PhMesiloxane capillary column. The mass detector was operated in the electron impact ionization mode (EIMS) with an electron energy of 70 eV. The injection port was heated to 250 °C. The oven temperature program was initially set at 70 °C with a hold of 1.5 min and ramped to 280 °C at 20 °C min−1 with a hold of 10 min. Electrospray ionization source-quadrupole time of flight (ESIQTOF) mass spectra, EIMS spectra, and 1H- and 13C NMR spectra identified correct and pure samples. Spectral data of all products agreed with those already reported for the same compounds prepared as previously reported.General procedure for the N-Fmoc and N-Cbz protection of amines 2a–s in [Bmim][BF4]
To a magnetically stirred mixture of amine 2a–s (1 mmol) and [Bmim][BF4] (1 mL) (for amines 2p–s 0.5 mL of NaHCO3 aqueous solution were added), Fmoc-OSu or Cbz-Osu (1 mmol) was added and the mixture was stirred at ambient temperature for 3–9 min. The reaction was monitored by TLC. After the completion of reaction, diethyl ether was added, and the IL settled at the bottom. The supernatant was decanted off and the IL was washed with Et2O (3 × 2 mL). The combined Et2O extracts were washed once with a 1 N aqueous solution of HCl (3 mL), dried over Na2SO4 and filtered. The products were isolated after evaporation of the diethyl ether to yield the highly pure N-Fmoc derivatives 3a–s in 85–97% yields or the N-Cbz-derivatives 4a–s in 85–95% yields. 1H NMR and 13C NMR were consistent with the assigned structures.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2), 5.83–5.95 (m, 1H, CH2CHCH2), 7.30–7.48 (m, 4H, ArHFmoc), 7.61 (d, J = 7.1 Hz, 2H, ArHFmoc), 7.79 (d, J = 7.1 Hz, 2H, ArHFmoc). 13C-NMR: (75 MHz, CDCl3) δ 43.5, 47.3, 66.7, 116.1, 120.0, 125.1, 127.1, 127.7, 134.5, 141.4, 144.0, 156.3. ESI(+)-MS: calcd for ([C18H17NO2] + Na)+ [M + Na]+ 302.1157; found: 302.1151.CH2), 5.82 (m, 1H, CHCH2), 7.38 (s, 5H, ArH). 13C-NMR: (75 MHz, CDCl3) δ 43.5, 66.8, 116.1, 127.9, 128.1, 128.5, 134.5, 136.6, 156.3. EIMS m/z (%) 191 (3, M+˙), 150 (2), 130 (16), 108 (68), 91 (100), 79 (15), 77 (14), 65 (7).
CHCH2), 5.83–5.95 (m, 1H, CH2CHCH2), 7.30–7.48 (m, 4H, ArHFmoc), 7.61 (d, J = 7.1 Hz, 2H, ArHFmoc), 7.79 (d, J = 7.1 Hz, 2H, ArHFmoc). 13C-NMR: (75 MHz, CDCl3) δ 43.5, 47.3, 66.7, 116.1, 120.0, 125.1, 127.1, 127.7, 134.5, 141.4, 144.0, 156.3. ESI(+)-MS: calcd for ([C18H17NO2] + Na)+ [M + Na]+ 302.1157; found: 302.1151.CH2), 5.82 (m, 1H, CHCH2), 7.38 (s, 5H, ArH). 13C-NMR: (75 MHz, CDCl3) δ 43.5, 66.8, 116.1, 127.9, 128.1, 128.5, 134.5, 136.6, 156.3. EIMS m/z (%) 191 (3, M+˙), 150 (2), 130 (16), 108 (68), 91 (100), 79 (15), 77 (14), 65 (7).Recycling of [Bmim][BF4] in the synthesis of 3h
At the end of the first experiment and after the recovery of the N-Fmoc-cyclopentylamine, the IL was treated with a saturated aqueous solution of NaHCO3 (2 mL) and subjected to consecutive extractions with ethyl acetate and diethyl ether. The ionic liquid was recovered by removing water under vacuum. The IL was finally dried at 80 °C for 1 h and reused for the next run. The IL was reused for consecutive four times without loss in its efficiency (91, 89, 88 and 86% yields, respectively).Acknowledgements
This work was supported by grants from MIUR (Ministero Italiano dell' Università e della Ricerca, ex-60% funds).Notes and references
- (a) P. G. M. Wuts and T. W. Green, Greene’s Protective Groups in Organic Synthesis, John Wiley and Sons, Hoboken, NJ, 4th edn, 2007 Search PubMed; (b) P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, New York, 3rd edn, 2005 Search PubMed; (c) M. L. Di Gioia, A. Leggio, A. Le Pera, A. Liguori, A. Napoli, F. Perri and C. Siciliano, J. Chromatogr. A, 2005, 1066, 143–148 CrossRef CAS PubMed.
- (a) E. Wuensch, Houben-Weyl, Methods of Organic Chemistry, ed., E. Muller, O. Bayer, H. Meerwin and K. Ziegler, George Thieme, Stuttgart, New York, 4th edn, 1974, vol. 15/1, p. 46 Search PubMed; (b) A. Leggio, M. L. Di Gioia, F. Perri and A. Liguori, Tetrahedron, 2007, 63, 8164–8173 CrossRef CAS PubMed; (c) A. Leggio, D. Alò, E. L. Belsito, M. L. Di Gioia, E. Romio, C. Siciliano and A. Liguori, J. Pept. Sci., 2015 DOI:10.1002/psc.2777.
- A. Ilangovan and R. G. Kumar, Chem.–Eur. J., 2010, 16, 2938–2943 CrossRef CAS PubMed.
- (a) F. Albericio, Biopolymers, 2000, 55, 123 CrossRef CAS; (b) L. A. Carpino, H. G. Chao, M. Beyermann and M. Bienert, J. Org. Chem., 1991, 56, 2635 CrossRef CAS; (c) L. A. Carpino, Acc. Chem. Res., 1987, 20, 401 CrossRef CAS; (d) M. L. Di Gioia, A. Leggio, A. Liguori, F. Perri, C. Siciliano and M. C. Viscomi, Amino Acids, 2010, 38, 133–143 CrossRef CAS PubMed; (e) M. L. Di Gioia, A. Leggio, A. Le Pera, C. Siciliano, A. Liguori and G. Sindona, J. Pept. Res., 2004, 63, 383–387 CrossRef CAS PubMed; (f) M. L. Di Gioia, A. Leggio, A. Le Pera, C. Siciliano, A. Liguori and G. Sindona Eur, J. Org. Chem., 2004, 21, 4437–4441 Search PubMed.
- (a) L. A. Paquette, in Encyclopedia of Reagents for Organic Synthesis, Wiley, Chichester, 1995, vol. 1, pp. 323–326 Search PubMed; (b) M. Bodanszky, Peptide Chemistry, Springer, Berlin, 1988 Search PubMed.
- (a) G. W. Anderson and A. C. McGregor, J. Am. Chem. Soc., 1957, 79, 6180–6183 CrossRef CAS; (b) R. A. Boissonnas, Advances in Organic Chemistry: Methods and Results, J. Wiley, 1963, vol. 3, p. 159 Search PubMed.
- (a) V. Perron, S. Abbott, N. Moreau, D. Lee, C. Penney and B. Zacharie, Synthesis, 2009, 283–289 CAS; (b) A. R. Katritzky, N. E. Abo-Dya, A. Abdelmajeid, S. R. Tala, M. S. Amine and S. A. El-Feky, Org. Biomol. Chem., 2011, 9, 596–599 RSC; (c) C. Helgen and C. G. Bochet, J. Org. Chem., 2003, 68, 2483–2486 CrossRef CAS PubMed; (d) R. Chinchilla, D. J. Dodsworth, C. Najera and J. M. Soriano, Bioorg. Med. Chem. Lett., 2002, 12, 1817–1820 CrossRef CAS; (e) R. Chinchilla, D. J. Dodsworth, C. Najera and J. M. Soriano, Tetrahedron Lett., 2001, 42, 7579–7581 CrossRef CAS; (f) B. H. Hu and P. B. Messersmith, Tetrahedron Lett., 2000, 41, 5795–5798 CrossRef CAS; (g) M. B. Gawande and P. S. Branco, Green Chem., 2011, 13, 3355 RSC; (h) M. Nardi, N. H. Cano, P. Costanzo, M. Oliverio, G. Sindona and A. Procopio, RSC Adv., 2015, 5, 18751–18760 RSC.
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1998, p. 531 Search PubMed; (b) V. Siddaiah, G. M. Basha, R. Srinivasarao and V. Yessayya, Green Chem. Lett. Rev., 2012, 337–342 CrossRef CAS PubMed; (c) P. P. Bora, K. Vanlaldinpuia, L. Rokhum and G. Bez, Synth. Commun., 2011, 41, 2674–2683 CrossRef CAS PubMed; (d) J. J. Shrikhande, M. B. Gawande and R. V. Jayaram, Tetrahedron Lett., 2008, 49, 4799–4803 CrossRef CAS PubMed.
- (a) C. Lutz, V. Lutz and P. Knochel, Tetrahedron, 1998, 54, 6385 CrossRef CAS; (b) Y. Basel and A. Hassner, J. Org. Chem., 2000, 65, 6368–6380 CrossRef CAS; (c) J. J. Reddy, P. S. Lakshmi, G. V. S. Sharma and P. R. Krishna, Tetrahedron Lett., 2004, 45, 6963–6965 CrossRef PubMed; (d) A. Heydari and S. E. Hosseini, Adv. Synth. Catal., 2005, 347, 1929–1932 CrossRef CAS PubMed; (e) A. K. Chakraborti and S. V. Chankeshwara, Org. Biomol. Chem., 2006, 4, 2769–2771 RSC; (f) S. V. Chankeshwara and A. K. Chakraborti, Tetrahedron Lett., 2006, 47, 1087–1091 CrossRef CAS PubMed; (g) G. Bartoli, M. Bosco, M. Locatelli, E. Marcantoni, M. Massaccesi, P. Melchiorre and L. Sambri, Synlett, 2004, 10, 1794–1798 CrossRef; (h) N. Suryakiran, P. Prabhakar, S. T. Reddy, K. Rajesh and Y. Venkateswarlu, Tetrahedron Lett., 2006, 47, 8039–8042 CrossRef CAS PubMed; (i) A. Heydari, R. K. Shiroodi, M. Esfandyari and M. Pourayoubi, Tetrahedron Lett., 2007, 48, 5865–5867 CrossRef CAS PubMed; (j) D. J. Upadhyaya, A. Barge, R. Stefania and G. Gravotto, Tetrahedron Lett., 2007, 48, 5865–5867 CrossRef PubMed; (k) G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199–250 CrossRef CAS PubMed.
- (a) P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, 2004 Search PubMed; (b) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2008, 108, 2015–2050 CrossRef CAS PubMed.
- (a) M. J. Earle and R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS; (b) J. D. Revell and A. Ganesan, Org. Lett., 2002, 4, 3071–3073 CrossRef CAS PubMed; (c) P. Cserjesi, K. Belafi-Bako, N. Nemestothy and L. Gubicza, Hung. J. Ind. Chem., 2008, 36, 27–34 Search PubMed; (d) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC; (e) Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- (a) J. Dupont and J. Spencer, Angew. Chem., Int. Ed., 2004, 43, 5296–5297 CrossRef CAS PubMed; (b) M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC; (c) J. Ranke, S. Stolte, R. Storman, J. Arning and B. Jastor, Chem. Rev., 2007, 107, 2183–2206 CrossRef CAS PubMed; (d) P. Dominguez de Maria, Angew. Chem., Int. Ed., 2008, 47, 6960–6968 CrossRef CAS PubMed; (e) D. M. Costello, L. M. Brown and G. A. Lamberti, Green Chem., 2009, 11, 548–553 RSC; (f) D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- M. L. Di Gioia, A. Barattucci, P. Bonaccorsi, A. Leggio, L. Minuti, E. Romio, A. Temperini and C. Siciliano, RSC Adv., 2014, 4, 2678–2686 RSC.
- (a) A. Sarkar, S. R. Roy, N. Parikh and A. K. Chakraborti, J. Org. Chem., 2011, 76, 7132–7140 CrossRef CAS PubMed; (b) S. Sunitha, S. Kanjilal, P. S. Reddy and R. B. N. Prasad, Tetrahedron Lett., 2008, 49, 2527–2532 CrossRef CAS PubMed; (c) M. A. Zolfigol, V. Khakyzadeha, A. R. Moosavi-Zarea, G. Chehardoli, F. Derakhshan-Panaha, A. Zare and O. Khalediana, Sci. Iran., Trans. C, 2012, 19, 1584–1590 CrossRef CAS PubMed; (d) A. Chinnappan, D. La and H. Kim, RSC Adv., 2013, 3, 13324–13328 RSC.
- N. Suryakiran, K. C. Mahesh, D. Ramesh, J. J. P. Selvam and Y. Venkateswarlu, Tetrahedron Lett., 2008, 49, 2607–2610 CrossRef CAS PubMed.
- R. Chinchilla, D. J. Dodsworth, C. Najera and J. M. Soriano, Bioorg. Med. Chem. Lett., 2001, 12, 1817–1820 CrossRef.
- P. B. W. Ten Kortenaar, B. G. Van Dijk, J. M. Peters, B. J. Raaben, P. J. H. M. Adams and G. I. Tesser, Int. J. Pept. Protein Res., 1986, 27, 398–400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copy of 1H and 13C NMR spectra for compounds 3a–o and 4a–o. See DOI: 10.1039/c5ra12121c |
| This journal is © The Royal Society of Chemistry 2015 |