
Synthesis, biological activity and structural study of new benzotriazole-based protein kinase CK2 inhibitors†
Abstract
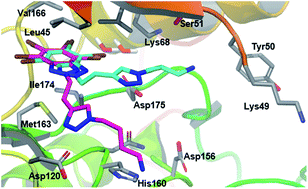
A new series of 4,5,6,7-tetrabromobenzotriazole (TBB) derivatives was synthesized and characterized as CK2 inhibitors. They were readily synthesized using a click chemistry approach based on a Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC). Some of the synthesized compounds present interesting inhibitory activities using an in vitro assay, with Ki values in the low micro molar range and a high degree of selectivity against a panel of 24 kinases. Selected compounds were tested for their antiproliferative effect on several cancer cell lines, and for their proapoptotic activity towards human Jurkat T-leukemia and MCF-7 breast adenocarcinoma cells, showing that they can be proposed as promising anticancer agents. Docking studies as well as crystallographic analysis allowed us to identify ligand–CK2 interactions that account for the molecular recognition process, and can help to further optimize this family of compounds as CK2 inhibitors.
 Please wait while we load your content...
Please wait while we load your content...

