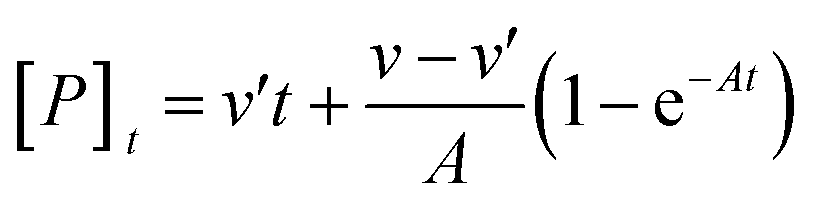DOI:
10.1039/C5RA12068C
(Paper)
RSC Adv., 2015,
5, 68906-68913
Determination of the kinetics and influence of the mercury ion on papain catalytic activity†
Received
23rd June 2015
, Accepted 30th July 2015
First published on 31st July 2015
Abstract
The effect of the mercury ion on papain activity of the substrate casein was investigated. Mercury ions (Hg2+) at low concentrations induced an increase of papain activity, but decreased it at high concentrations, confirming a typical hormesis phenomenon. Papain activity increased to a maximum of 111.03% at a concentration of 10−6 mol L−1 Hg2+, but was almost completely deactivated at concentrations above 10−4 mol L−1 Hg2+. The conformational changes in papain structure because of the interaction of Hg2+ and papain were studied using synchrotron radiation circular dichroism, attenuated total reflectance Fourier-transform infrared and intrinsic fluorescence spectroscopies, and the catalytic behavior of the enzyme was studied using kinetic analysis. At up to 10−4 mol L−1 concentrations, Hg2+ significantly decreased the α-helix content of papain and increased the random coil content so that the papain with a lower affinity to substrate was nearly completely inactivated. However, papain activity increased with an increase of the α-helix content and decrease of random coils when the Hg2+ concentration at 10−6 mol L−1. There were different modification mechanisms of papain activity with different concentrations of Hg2+. At a concentration of 10−6 mol L−1 Hg2+, Hg2+ was acting as an efficacious activator, and the impact could be classified as a non-competitive type. At a concentration of 10−4 mol L−1 Hg2+, the inhibition of Hg2+ on papain was found to be a competitive and uncompetitive mixed type, and Hg2+ at this concentration bound to the enzyme molecule leading to the loss of enzyme activity. As a result, the detection limit of papain was 10−4 mol L−1 and has a potential application for determining low doses of Hg2+.
1. Introduction
Because of the rapid and unceasing industrial development in China, heavy metal contaminants have been introduced into natural waters, soil and air. These metal contaminants, unlike organic pollutants, cannot be detoxified via degradation and thus persist in the ecosystem. They get into the human food chain from the environment causing hazardous effects on human, animal and plant organisms.1–3 Certain metal ions are highly toxic, and the determination of traces of toxic heavy metals in these environmental pollutants has become very important. Among them, mercury ions (Hg2+) have attracted the most attention because of its strong toxicity and the increasing level of its extended use in industrial processes.4–6
Mercury can be determined by atomic absorption spectrometry, X-ray fluorescence spectroscopy, high performance liquid chromatography and electrochemical methods. The disadvantages of these methods are the complicated operation process and expensive equipment and the need for skilled operators.7 Heavy metals are well known as inhibitors of enzyme activity and the application of this phenomenon to the determination of these hazardous toxic elements using enzymes has received considerable attention and offers several advantages, such as relatively short response time, high sensitivity, selectivity and specificity. Most of these enzymes are cheap and do not require costly instruments or have stringent requirements for use as toxic substance bioassays do. Thus, a lot of enzymes have been used for the inhibitive determination of trace heavy metal ions in environmental samples. Some enzymes such as papain (EC 3.4.22.2), urease (EC 3.5.1.5), glucose oxidase (EC 1.1.3.4), and xanthine oxidase (EC 1.17.3.2) have been used for the determination of Hg2+.1,3,4,7–10 Because mercury is a soft acid and the residues containing thiol group are soft bases, mercury targets the thiol containing enzymes, irreversibly binding their critical thiol groups, consequently leading to an inactivation of the enzyme.11–13 So the inhibition of enzymic activity by mercury may be a good choice as a simple, rapid and sensitive screening test.
Papain (EC 3.4.22.2) is a highly stable enzyme, one of the proteolytic enzymes obtained from papaya latex.14 Papain is a cysteine protease consisting of 212 amino acid residues including seven Cys residues and is stabilized by three disulfide bridges.15,16 Hg2+ (soft acid) has a strong bond with the cysteine residues in papain (soft base) resulting in irreversible inhibition of papain.11,17,18 The papain assay has a wide pH for optimum activity, temperature stability and sensitiveness to heavy metals so that the papain assay is used to monitor heavy metal concentrations, including Hg2+, in water environments.3,13 When enzymes are exposed for minutes to Hg2+, there is an influence on the activity of enzymes such as tyrosine kinase, phospholipase C and Ca2+-ATPase.19,20
Hormesis is the rule rather than an exception, and it represents an evolutionary-based adaptive response to environmentally induced disruption in homeostasis. Although the biphasic dose-response is a common result of experiments, the low dose data have been largely ignored. The hormesis phenomenon, low dose stimulation followed by high dose inhibition, is relatively commonly observed among classes of organisms including enzyme activity in response to various heavy metals.21,22 Hg2+ is one of the most hazardous heavy metals in the environment. Most of the studies performed have been more concerned about the toxic effects of Hg2+ at high concentrations, and much less information is available about the effect of Hg2+ at low concentrations. For determining traces of mercury in water, it is essential to investigate enzyme activity in response to different Hg2+ concentrations.
The main aim of the present work was to evaluate the influence of Hg2+ on papain activity. The combination of synchrotron radiation circular dichroism (SRCD), attenuated total reflectance – Fourier-transform infrared (ATR-FTIR) and intrinsic fluorescence spectroscopies as well as kinetics analysis were used to understand the structure–function relationship of papain in the presence of Hg2+. The present work is useful for understanding the interaction mechanism that takes place between mercury and papain and it will be used to create potential applications of papain as a bioindicator for heavy metals.
2. Materials and methods
2.1. Enzyme and reagents
Papain (≥99%), bovine serum albumin, tyrosine and casein were purchased from Sigma-Aldrich (Shanghai, China). All other reagents used were of analytical grade and used without further purification. All solutions were made with redistilled and ion free water.
2.2. Effect of Hg2+ on papain activity
Papain activity was measured as described by Guo et al.23 Papain solution (1.0 mg mL−1) was obtained by dissolving the enzyme in phosphate buffered saline (PBS) buffer (0.1 mol L−1, pH 7.0). A stock solution of mercury(II) chloride (0.1 mol L−1) was prepared in the PBS buffer and it was diluted to the concentration required from 10−9 to 10−2 mol L−1 for the assays of papain activity in the presence of Hg2+. In the preliminary experiment, it was found that the reaction attained equilibrium after 30 min (ESI, Fig. S1†) which was the same as the equilibrium time obtained by Guo et al.23 So, the reaction time for the hydrolysis was set to 30 min. The papain solutions in the buffer were added initially at different Hg2+ concentrations. After 10 min at 40 °C, 3.0 mL of casein solution (2.0 mg mL−1) was added into the mixture at 40 °C for 30 min before the addition of 2.0 mL of 20% (by mass) trichloroacetic acid to stop the reaction. The activity of papain was determined using a U-9100 spectrophotometer (Hitachi, Tokyo, Japan) at 275 nm. One unit of enzyme activity was defined as 1 μg tyrosine formed per minute at 40 °C and pH 7.0. The relative activity (%) was the ratio of the enzyme activity in the PBS buffer at different Hg2+ concentrations and in the PBS buffer without Hg2+.
2.3. ATR-FTIR and intrinsic fluorescence spectroscopies
ATR-FTIR spectra of the samples in the ATR cells were recorded on a Spectrum One B instrument (PerkinElmer, Waltham, MA, USA). The background was subtracted using Opus software. Curve fitting was performed using Origin 9.0 and PeakFit v4.12 software. The tryptophan (Trp) fluorescence spectra were recorded using a LS55 spectrofluorimeter (PerkinElmer, Waltham, MA, USA) at 25 °C. The emission spectra were recorded in the range of 300–410 nm at 500 nm min−1, 10 s after excitation, keeping the excitation constant at 288 nm, with slit widths of 5 nm for excitation and emission. Tryptophan ethyl ester was used as an internal standard to correct the inner filter effect. The blank spectra without enzyme was subtracted from the sample spectra.
Papain (0.5 mg mL−1) was equilibrated in the solutions with 0 (control), 10−6, 10−5 or 10−4 mol L−1 concentrations of Hg2+ at 40 °C for 10 min, and then centrifuged at 3000 rpm (equal to a g value of 800) for 4 min. The supernatant was used for ATR-FTIR and fluorescence spectral measurements. Triplicate samples were analyzed and spectra for the triplicate runs were averaged and used as the final spectral data.
2.4. Synchrotron radiation circular dichroism (SRCD) spectroscopy
Samples were examined in circular demountable 0.0015 cm path length Suprasil cells (Hellma, Southend on Sea, UK), which had been previously calibrated using interferometry methods.24 At the 4B8 beam line of the Beijing Synchrotron Radiation Facility (Beijing, China), three repeat measurements of each of the protein samples were made over the wavelength range from 280 to 165 nm at 5 °C, using a 1 nm interval and a time constant of 5 s. At CD1, spectra were measured using an interval of 1 nm and dwell time of 2.1 s. Five repeat measurements of each protein spectrum were measured from 260 to 172 nm at 25 °C. Spectral data from both beam lines were processed using identical procedures with CD tool software, and secondary structure analyses were performed using CDPro software package (at http://lamar.colostate.edu/~sreeram/CDPro/main.html), which consisted of three of the popular programs (SELCON3, CDSSTR and CONTINLL) for analyzing the protein CD spectra to determine the secondary structure fractions.25,26 The fitting was then performed using the three programs, and the best fitting procedure was based on root-mean-square deviation [RMSD(Exp-Calc)] and normalized root mean squared deviation [NRMSD(Exp-Cal)].
2.5. Kinetic measurements
The effect of the mercury was caused by the tight binding of mercury to a reactive –SH group in the enzyme, and the effect was irreversible.11,12,17 In order to investigate the irreversible modification by Hg2+ on papain activity, the kinetic model of substrate reaction during irreversible modification of enzyme activity described by Tsou was used to study the kinetics of papain by Hg2+. The model was not only suitable for inhibition kinetics, but also for activation kinetics.27–29 The kinetic method described by Tsou had been used in studies of inactivation of various enzymes by inhibitors.30–32 However, most of the studies were focused on single inhibition30–32 or single activation,33–35 but there has been little research on both inactivation and activation of an enzyme by a modificator. In this research, the irreversible modification (inhibition/activation) of Hg2+ on papain activity was investigated. The reaction mechanism is shown in Scheme 1, where E, S, P and Y represent papain, substrate casein, product tyrosine and Hg2+, respectively. EY, ES and EYS are the respective complexes.
 |
| | Scheme 1 Modification of papain by Hg2+ in the presence of substrate. | |
As is usual, the case [S] ≫ [E0] and the modification reactions were relatively slow compared with the set-up of the steady state of the enzymic reaction. The concentration of the product formed can be written as:29
| |
 | (1) |
| |
 | (2) |
where [
P]
t is the concentration of the product formed at the reaction time
t,
A is the apparent rate constant, [
S] is the concentration of casein,
v and
v′ are the reaction velocities of the reaction in the absence and presence of Hg
2+ at time
t, respectively,
Km and
K′m are the Michaelis constants,
k0 and
k′0 are the dissociation constants for the modifier with different forms of the enzyme, respectively, and
Vm and
V′m are the maximum reaction velocities. When
v >
v′, the modifier Hg
2+ was an inhibitor. When
v <
v′, the modifier Hg
2+ was an activator. When the reaction time
t was sufficiently long, the curves become straight lines and the product concentration was written as [
Pe]:
| |
 | (3) |
3. Results and discussion
3.1. Effect of Hg2+ on papain activity
The effect of Hg2+ concentration on papain activity was investigated and typical dose response phenomenon (hormesis) characterized by low dose stimulation and high dose inhibition is shown in Fig. 1. Hg2+ inhibited papain activity with a relative activity of 6.81% when Hg2+ concentration was ≥10−4 mol L−1, but it was observed that stimulation of papain activity could occur at an Hg2+ concentration of 10−6 mol L−1 and displayed the highest relative activity of 111.03%. There was no significant difference in papain activity, when exposed to concentrations of 10−10–10−7 and 10−5 mol L−1 of Hg2+ buffer. In order to see the effect of chloride ions, the effect of potassium chloride on the enzyme activity was checked and there was no change in enzyme activity (data not shown). Thus, the change in the activity observed was mainly because of the Hg2+ alone. According to the results of the experiment, three different concentrations of Hg2+, including 10−6, 10−5 and 10−4 mol L−1, were chosen to investigate the interactions between Hg2+ and papain activity.
 |
| | Fig. 1 Effect of Hg2+ concentration on papain activity. | |
3.2. Influence of Hg2+ on the secondary structure of papain
It was possible that the change in enzyme activity observed in the buffer containing metal ions might be because of the papain secondary structure changes. In the preliminary work, it was found, using the spectra of ATR-FTIR and intrinsic fluorescence spectroscopy, that there was little difference between native papain and the papain samples exposed to 10−7 and 10−5 mol L−1 Hg2+ buffer. So, the changes in the secondary structure of papain as a function of metal ion concentration were determined by ATR-FTIR, intrinsic fluorescence and SRCD spectroscopies in the presence of the 10−6, 10−5 and 10−4 mol L−1 Hg2+ concentrations.
3.3. ATR-FTIR
ATR-FTIR spectroscopy has been used extensively to study the changes in the secondary structure of protein. The amide I band between 1700–1600 cm−1 was the most useful for spectroscopic analysis of the secondary structure of protein.36 The original ATR-FTIR spectra of the individual papain sample in the 1800–900 cm−1 region were measured and the component peaks in the amide I region were determined using the curve fitting method and the results are shown in Fig. 2 and Table 1, in which the individual component locations of the papain secondary structure were assigned according to the methods used in earlier studies.36–38 It was found that Hg2+ had an effect on the secondary structure of papain. Compared with the control, the papain exposed to 10−6 mol L−1 Hg2+ buffer exhibited an increase in α-helix and β-sheet content and a decrease in β-turn and random coil content, and the percentage of the aggregated structure in the papain was decreased from 8.40% to 5.33%. Again, there was no significant change in the structural features of the papain exposed to 10−5 mol L−1 Hg2+ buffer, implying that no major conformational changes occurred in the papain. However, in the presence of 10−4 mol L−1 Hg2+, the α-helix content of the enzyme significantly decreased from 34.57% to 18.47%. The β-turn and random coil content rose from 19.19% to 22.38% and 23.47% to 31.69%, respectively. In particular, the papain dramatically decreased the contribution of the α-helix and increased the aggregated structure, but there was a very small decrease in β-sheet content from 14.37% to 11.11%. The results showed that the enzyme activity increased with the increase of α-helix content and decrease of the intermolecular β-sheet aggregate and random coil contents.
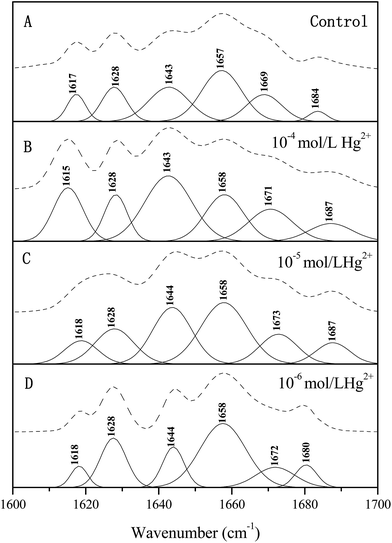 |
| | Fig. 2 Original FTIR spectra and individual Gaussian bands of papain in amide I region original FTIR spectra (upper); – individual Gaussian bands (bottom) (a) control; (b) 10−4 mol L−1 Hg2+; (c) 10−5 mol L−1 Hg2+; (d) 10−6 mol L−1 Hg2+. | |
Table 1 Secondary structure areas and assignments in amide I infrared bands of papain in different Hg2+ concentrationsa
| Second structure |
Hg2+ concentration (mol L−1) |
| Control |
10−6 |
10−5 |
10−4 |
| Peak centers (cm−1) |
Area (%) |
Peak centers (cm−1) |
Area (%) |
Peak centers (cm−1) |
Area (%) |
Peak centers (cm−1) |
Area (%) |
| The reactions were performed at 40 °C, pH 7.0 for 30 min, 0.5 mg mL−1 papain Tris–HCl solution was equilibrated in 0 (control), 10−6, 10−5 and 10−4 mol L−1 concentrations of Hg2+ at 25 °C for 10 min, and then centrifuged at 3000 rpm (equal to a g value of 800) for 4 min. The supernatant was used to determine the structure changes of the treated papain. The papain without Hg2+ treatment was used as the control. All the samples were analysed three times and the data obtained from the triplicate runs were averaged and used as the final result. The contribution of the individual components of the secondary structure of papain in Hg2+-PBS buffer was based on the research reported by Llerena-Suster et al.39 |
| Intermolecular β-sheet aggregates |
1617 |
8.40 |
1618 |
5.33 |
1618 |
8.62 |
1615 |
16.35 |
| β-Sheet |
1628 |
14.37 |
1628 |
18.96 |
1628 |
15.15 |
1628 |
11.11 |
| α-Helix |
1657 |
34.57 |
1658 |
43.73 |
1658 |
31.46 |
1658 |
18.47 |
| Random coil |
1643 |
23.47 |
1644 |
12.95 |
1644 |
24.44 |
1643 |
31.69 |
| β-Turn |
1669 |
19.19 |
1672 |
19.03 |
1673 |
20.33 |
1671 |
22.38 |
| 1684 |
1680 |
1687 |
1687 |
3.4. Intrinsic fluorescence spectroscopy
Intrinsic fluorescence spectroscopy was used to investigate the perturbation of Trp residues in papain as a result of Hg2+ interaction with enzyme, and the fluorescence emission spectra of papain is shown in Fig. 3. Native papain in aqueous buffer showed a typical Trp peak with the maximum emission at about 342 nm and the fluorescence was mainly because of the presence of five Trp residues in papain,39 and the maximum emission of the control was at 340.5 nm (Fig. 3a). Upon interaction of papain with a Hg2+ concentration of 10−6 mol L−1, the maximum fluorescence emission appeared to show a slightly blue shift (2 nm, Fig. 3c) with a decrease in intensity compared to the control. This blue shift could be attributed to the conformational changes in the vicinity of the surface exposed Trp residues, presumably because of internalization in a more hydrophobic environment. For the papain exposed to 10−5 mol L−1 Hg2+ buffer, the emission maximum did not alter and there was a small decrease in intensity, implying that the papain basically maintained its native state in a 10−5 mol L−1 Hg2+ concentration (Fig. 3b). For a concentration of 10−4 mol L−1 Hg2+, there was a marked decrease in the emission intensity and a red shift of 7 nm in the emission maximum was found (Fig. 3d). It might be deduced that the internalized Trp residues in the native state were partially exposed from a hydrophobic to a hydrophilic environment leading to partial unfolding of the molecule. In the latter case, the Trp residues were enclosed inside a more polar protein, which resulted in an inactive enzyme. All these data clearly suggested that the structural alteration of papain exposed to Hg2+ buffers induced the microenvironmental change of the Trp residues.
 |
| | Fig. 3 Fluorescence emission spectra of papain exposed to different Hg2+ concentrations (a) control; (b) 10−5 mol L−1 Hg2+; (c) 10−6 mol L−1 Hg2+; (d) 10−4 mol L−1 Hg2+. | |
3.5. SRCD spectroscopy
Far ultraviolet (UV)-CD spectra of papain in the presence of Hg2+ concentrations at 10−6, 10−5 and 10−4 mol L−1 are shown in Fig. 4. The spectrum of native papain (control) had a negative trough at 208 nm and a shoulder at 220 nm (Fig. 4a). The effect of Hg2+ on papain was accompanied by only small change in the CD spectra of papain at low Hg2+ concentrations (≤10−5 mol L−1), whereas a larger change in the conformation of the papain molecule was observed at higher Hg2+ concentrations (≥10−4 mol L−1). All these signals were abolished after the papain was exposed to 10−4 mol L−1 Hg2+ buffer, indicating the disruption of the native secondary structure and a loss of the rigid tertiary structure. The secondary structural features of the samples are summarized in Table 2. The change of each secondary structural content of papain detected using SRCD spectroscopy analysis matched with those determined using ATR-FTIR spectroscopy, indicating that the change tendency at different Hg2+ concentrations was defined and verified with each other. Deformation in the papain secondary structure led to the change of the protein's native three-dimensional structure wherein the function of the protein could also be altered. Far UV-CD spectra revealed that the papain-Hg2+ complex structure has a certain deformation leading to the structural and functional change which might be considered as a significant factor in influencing its activity. The RMSD and NRMSD values fitted using IBasis6 in the SELCON3 program were smaller than 0.1, which indicated that the fitting method was well suited to the secondary structure analyses of papain samples.18,40,41 The α-helix content of the native papain molecule was about 43.4% which was obtained from the CD spectrum data. Of the two domains in the papain folding structure, the first domain of native papain had a large α-helix content, whereas the secondary domain was mainly β-sheet and a lesser amount of α-helix.42 Accompanied by slight enhancement of papain activity (Fig. 1), the papain in the presence of 10−6 mol L−1 Hg2+ presented a modest increase of α-helix content; furthermore, there was a slight decrease of the β-turn and random coil contents because of the binding of Hg2+. The Hg2+ at 10−5 mol L−1 concentration had almost no effect on the secondary structural contents of papain. Nevertheless, with exposure to 10−4 mol L−1 Hg2+, the binding of Hg2+ to papain dramatically decreased the α-helix content and significantly increased the β-sheet and random coil content leading to enzyme inactivation, whereas the β-turn changed very slightly compared with the native papain. The binding of Hg2+ to papain was marked by significant changes in the shape and position of the far UV-CD spectra.
 |
| | Fig. 4 Far UV-CD spectra of papain exposed to different Hg2+ concentrations (a) control; (b) 10−5 mol L−1 Hg2+; (c) 10−6 mol L−1 Hg2+; (d) 10−4 mol L−1 Hg2+. | |
Table 2 Secondary structure contents of papain in different Hg2+ concentrations, measured using CD spectroscopy
| Hg2+ concentration (mol L−1) |
Secondary structurea (%) |
| H(r) |
H(d) |
α-helix |
S(r) |
S(d) |
β-sheet |
β-turn |
Unrd |
| The secondary structure fraction results of papain were obtained using the SELCON3 program with IBasis6 at the wavelength range of 185–240 nm. The secondary structures are: H(r): regular α-helix; H(d): distorted α-helix; S(r): regular β-sheet; S(d): distorted β-sheet; Unrd: random coil. |
| Control |
28.2 |
15.2 |
43.4 |
5.3 |
7.4 |
12.7 |
16.0 |
27.2 |
| 10−6 |
32.2 |
16.3 |
48.5 |
4.3 |
6.1 |
10.4 |
15.0 |
26.1 |
| 10−5 |
27.4 |
14.5 |
41.9 |
5.4 |
7.4 |
12.8 |
17.1 |
28.2 |
| 10−4 |
1.8 |
2.4 |
4.2 |
27.8 |
12.2 |
40.0 |
19.6 |
36.2 |
As suggested previously, the results further confirmed that the enzyme activity increased with the increase of α-helix content as well as a decrease of random coil contents, and vice versa. It is important to note that papain activity could be influenced not just by active site geometry but also by the domain packing property. By virtue of a similar enzyme activity and the secondary structure to native papain, the papain exposed to 10−5 mol L−1 Hg2+ buffer was eliminated for further kinetic experiments.
3.6. Kinetic constants
The kinetic parameters, Km and Vmax, of the papain were calculated from the Lineweaver–Burk and Michaelis–Menten models in presence of different Hg2+ concentrations (10−6 and 10−4 mol L−1) and substrate casein concentrations (0.2, 0.4, 0.6, 0.8, 1.0, 1.6 and 2.0 mg mL−1) concentrations. Lineweaver–Burk reciprocal plots of the samples are shown in Fig. S2 (ESI†). For the papain exposed to 10−6 mol L−1 Hg2+ buffer, the Vmax value increased from 0.1281 mg min−1 for the control to 0.1400 mg min−1, while Km decreased from 2.3247 mg mL−1 for the control to 0.9144 mg mL−1. The decrease in Km indicated that the papain exposed to a low dose of Hg2+ (10−6 mol L−1) had an apparent higher affinity for its substrate than that of the control, and the Vmax value was therefore greater than that of the control. Tables 1 and 2 show that Hg2+ at a concentration of 10−6 mol L−1 increased the α-helix content of the papain, and this seemed to be related to the decrease of the random coil for the remaining structure contents. On the contrary, the Vmax value of the papain exposed to the 10−4 mol L−1 Hg2+ buffer was reduced to 0.0864 mg min−1, whereas Km increased to 2.4288 mg mL−1. Hg2+ at a concentration of 10−4 mol L−1 was able to weaken the affinity for the substrate by decreasing the α-helix content and enhancing the random coil content. These results suggested that the conformational change induced by Hg2+ led to a change in the affinity of the papain for the substrate.
3.7. Kinetics properties of papain in the presence of Hg2+
The time course of the hydrolysis of the substrate at concentrations of 10−6 and 10−4 mol L−1 Hg2+ are shown in Fig. 5. For the substrate hydrolysis in the presence of Hg2+, the rate increased with increasing substrate concentration, whereas the slope of the asymptote increased with increasing substrate concentration. The reaction progress curves of the control were linear over a lengthy period of time. The results were analyzed using Tsou's method29 and they suggested that Hg2+ at a concentration of 10−6 mol L−1 had a stimulating effect on enzyme activity, but a Hg2+ concentration of 10−4 mol L−1 had an inhibitory effect. According to eqn (1), the exponential linearized expressions were achieved using the least square fitting method of all the data shown in Fig. 5. The kinetic parameters of the model are shown in Table 3. The results showed that the calculated correlation coefficients (R2) were greater than 0.9891, indicating that the kinetic model could well describe the substrate hydrolysis during papain binding to Hg2+. According to eqn (3), the plot of 1/[Pe] against 1/[S] gave a straight line. The values of k0 and k′0 were calculated, and are given in Table 3.
 |
| | Fig. 5 Time course of the casein hydrolysis reaction by papain at different concentrations of Hg2+ and substrate A control; B 10−6 mol L−1 Hg2+; C 10−4 mol L−1 Hg2+. ⋯: Prediction; ■: experimental 1: 0.2 mg mL−1 casein; 2: 0.4 mg mL−1 casein; 3: 0.6 mg mL−1 casein; 4: 0.8 mg mL−1 casein; 5: 1.0 mg mL−1 casein. | |
Table 3 Kinetic parameters and dissociation constant of papain in casein hydrolysisa
| Hg2+ concentration (mol L−1) |
Casein concentration (mg mL−1) |
A (mg mL−1) |
ν′ (mg min−1) |
v (mg min−1) |
[Pe] (mg mL−1) |
R2 |
k0 |
k′0 |
| The reactions were performed at 40 °C, pH 7.0 for 30 min, with 1.0 mg mL−1 papain Tris–HCl solution in different concentrations of Hg2+ ion. Every group test was run three times and the mean values were used as the final test results. |
| Control |
0.2 |
— |
0.0031 |
0.0031 |
0.1967 |
0.9891 |
— |
— |
| 0.4 |
— |
0.0062 |
0.0062 |
0.3820 |
0.9951 |
| 0.6 |
— |
0.0092 |
0.0092 |
0.5640 |
0.9953 |
| 0.8 |
— |
0.0100 |
0.0100 |
0.7050 |
0.9952 |
| 1.0 |
— |
0.0105 |
0.0105 |
0.8300 |
0.9959 |
| 10−6 |
0.2 |
0.0092 |
0.0124 |
0.0031 |
0.1984 |
0.9971 |
0.1509 |
0.0150 |
| 0.4 |
0.0153 |
0.0182 |
0.0062 |
0.3878 |
0.9990 |
| 0.6 |
0.0143 |
0.0280 |
0.0092 |
0.5793 |
0.9994 |
| 0.8 |
0.0136 |
0.0235 |
0.0100 |
0.7604 |
0.9996 |
| 1.0 |
0.0081 |
0.0590 |
0.0105 |
0.8843 |
0.9999 |
| 10−4 |
0.2 |
0.0334 |
0.0030 |
0.0031 |
0.1069 |
0.9996 |
0.0634 |
0.0344 |
| 0.4 |
0.0503 |
0.0059 |
0.0062 |
0.2123 |
0.9997 |
| 0.6 |
0.0643 |
0.0089 |
0.0092 |
0.2994 |
0.9994 |
| 0.8 |
0.0885 |
0.0099 |
0.0100 |
0.3645 |
0.9990 |
| 1.0 |
0.1004 |
0.0104 |
0.0105 |
0.4509 |
0.9989 |
The v value of native papain was less than the v′ value of papain exposed to 10−6 mol L−1 Hg2+ buffer, indicating that the Hg2+ could stimulate papain activity. Furthermore, the values of the dissociation constants k0 and k′0 could be obtained, and the value of k0 (0.1509) was 10 times as much as that of k′0 (0.0150). This showed that there was a binding of Hg2+ to both native enzyme (E) and enzyme–substrate complex (EY). The binding of Hg2+ to papain had two existing forms, EY and EYS, and EYS was greater than EY. The apparent rate constant A was shown to be independent of [S] using the data in Table 3, implying that Hg2+ at a concentration of 10−6 mol L−1 was a non-competitive activator for the enzyme and activation had nothing to do with substrate concentration. The results strongly indicated that the amino acid residues responsible for the binding interactions of Hg2+ to the enzyme were mainly located outside the active center of papain and induced the small conformation change of the residues leading to an increase of enzyme activity.
At a concentration of 10−4 mol L−1 Hg2+, the v value was higher than the v′ value, revealing that Hg2+ was an inhibitor for papain. The values of k0 (0.0634) and k′0 (0.0344) showed that the Hg2+ binding of papain had two existing forms, EY and EYS. A plot of 1/A versus [S] gave a nearly linear slope (R2 = 0.9938), and the A value increased with increasing substrate concentration [S], implying that Hg2+ at a concentration of 10−4 mol L−1 was a competitive inhibitor for papain. At the same time, it could be seen that Hg2+ at a concentration of 10−4 mol L−1 could combine with both native enzyme (E) and enzyme–substrate complex (ES) for forming EY and EYS, respectively, and it displayed a competitive and uncompetitive mixed type mechanism. From k0 ≈ 2k′0, it was found that the binding of Hg2+ to papain mainly occurred at the amino acid residues from the active site of papain, and others outside the active site.
4. Conclusion
This study demonstrated that there were significant impacts of Hg2+ on papain, where the papain activity in the hydrolysis of casein was increased by Hg2+ at low concentrations but inhibited at high concentrations, which indicated the occurrence of a hormetic phenomenon. The strongest inhibition effect was observed when the Hg2+ concentration was up to 10−4 mol L−1, whereas mild activation was 10−6 mol L−1. So, the lowest detection limit for papain was a 10−4 mol L−1 Hg2+ concentration. The results showed that papain was suitable for the determination of Hg2+ in environmental analysis. The fundamental correlations between the structure of papain and its activity were clarified. The three-dimensional structure of the papain exposed to Hg2+ buffers was determined using ATR-FTIR, SRCD and intrinsic fluorescence spectroscopies. The papain exposed to 10−6 mol L−1 Hg2+ buffer had an increase in α-helix content and a decrease in random coil content, but the papain exposed to a 10−4 mol L−1 Hg2+ buffer had an increase in random coil content but a decrease in α-helix content. The enzyme activity increased with the increase of α-helix content and decrease of random coil contents, and vice versa. At a concentration of 10−6 mol L−1 Hg2+, the binding sites of the modifier with papain were basically situated outside of the active site of papain, where Hg2+ might induce a change of enzyme conformation leading to an increase in the affinity for the substrate and papain activity. At a concentration of 10−4 mol L−1 Hg2+, the modifier bonded with the amino acid residues within and outside the active center of papain, and Hg2+ shifted the three-dimensional structure of papain which was conducive to a decrease in affinity for substrate and occurrence of strong inhibition of the papain activity. This indicated that there was difference between the modification mechanisms of Hg2+ on papain activity at concentrations of 10−4 mol L−1 and 10−6 mol L−1.
Acknowledgements
This work is supported by National Natural Science Foundation of China (21105085, 31270988), the Hunan Provincial Natural Science Foundation of China (No. 2015JJ2133), the Scientific Research Fund of the Hunan Provincial Education Department (13B120) and the Economical Forest Cultivation and Utilization of 2011 Collaborative Innovation Center in Hunan Province [(2013) 448].
References
- M. R. Guascito, C. Malitesta, E. Mazzotta and A. Turco, Sens. Actuators, B, 2008, 131, 394–402 CrossRef CAS PubMed.
- C. M. Jonsson, L. C. Paraiba and H. Aoyama, Ecotoxicology, 2009, 18, 610–619 CrossRef CAS PubMed.
- Y. Shukor, N. A. Baharom, F. A. Rahman, M. P. Abdullah, N. A. Shamaan and M. A. Syed, Anal. Chim. Acta, 2006, 566, 283–289 CrossRef CAS PubMed.
- F. Kuralay, H. Özyörük and A. Yıldız, Enzyme Microb. Technol., 2007, 40, 1156–1159 CrossRef CAS PubMed.
- P. Mahato, S. Saha, P. Das, H. Agarwalla and A. Das, RSC Adv., 2014, 4, 36140–36174 RSC.
- M. Govindhan, B.-R. Adhikari and A. Chen, RSC Adv., 2014, 4, 63741–63760 RSC.
- Y. Yang, Z. Wang, M. Yang, M. Guo, Z. Wu, G. Shen and R. Yu, Sens. Actuators, B, 2006, 114, 1–8 CrossRef CAS PubMed.
- T. Krawczyński vel Krawczyk, M. Moszczyńska and M. Trojanowicz, Biosens. Bioelectron., 2000, 15, 681–691 CrossRef.
- M. S. Mondal and S. Mitra, J. Inorg. Biochem., 1996, 62, 271–279 CrossRef CAS.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473–4491 RSC.
- M. J. Ramírez-Bajo, P. de Atauri, F. Ortega, H. V. Westerhoff, J. L. Gelpí, J. J. Centelles and M. Cascante, PloS one, 2014, 9, e80018 Search PubMed.
- R. Sharma, in Enzyme Inhibition and Bioapplications, ed. R. R. Sharma, InTech, Rijeka, 2012, DOI:10.5772/39273.
- M. Shukor, N. Masdor, N. Baharom, J. Jamal, M. Abdullah, N. A. Shamaan and M. Syed, Appl. Biochem. Biotechnol., 2008, 144, 283–291 CrossRef CAS.
- H.-L. Nie, T.-X. Chen and L.-M. Zhu, Sep. Purif. Technol., 2007, 57, 121–125 CrossRef CAS PubMed.
- E. Amri and F. Mamboya, Am. J. Biochem. Biotechnol., 2012, 8, 99–104 CrossRef CAS.
- P. Kaul, H. Sathish and V. Prakash, Food Nahrung, 2002, 46, 2–6 CrossRef CAS.
- I. J. Kade, J. Biomed. Biotechnol., 2012, 2012, 924549 Search PubMed.
- P. Pancoska and T. A. Keiderling, Biochemistry, 1991, 30, 6885–6895 CrossRef CAS.
- B. Burlando, M. Bonomo, E. Fabbri, F. Dondero and A. Viarengo, Cell Calcium, 2003, 34, 285–293 CrossRef CAS.
- B. Burlando, V. Magnelli, I. Panfoli, E. Berti and A. Viarengo, Cell. Physiol. Biochem., 2003, 13, 147–154 CrossRef CAS PubMed.
- C. R. Wang, Y. Tian, X. R. Wang, H. X. Yu, X. W. Lu, C. Wang and H. Wang, Chemosphere, 2010, 80, 965–971 CrossRef CAS PubMed.
- Y. Zhang, G. Shen, Y. Yu and H. Zhu, Environ. Pollut., 2009, 157, 3064–3068 CrossRef CAS PubMed.
- Y. Guo, Z. Wang, W. Qu, H. Shao and X. Jiang, Biosens. Bioelectron., 2011, 26, 4064–4069 CrossRef CAS PubMed.
- A. J. Miles, F. Wien, J. G. Lees and B. Wallace, Spectroscopy, 2005, 19, 43–51 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, Methods Enzymol., 2004, 318–350 CAS.
- B. Wallace, J. Lees, A. Orry, A. Lobley and R. W. Janes, Protein Sci., 2003, 12, 875–884 CrossRef CAS PubMed.
- P. B. Chock, C. Huang, C. Tsou and J. Wang, Journal, 1988, 342–350 Search PubMed.
- A. G. McDonald and K. F. Tipton, eLS, 2012 Search PubMed.
- C. Tsou, Adv. Enzymol. Relat. Areas Mol. Biol., 1988, 61, 381–436 CAS.
- Q.-X. Chen, Z. Zhang, X.-W. Zhou and Z.-L. Zhuang, Int. J. Biochem. Cell Biol., 2000, 32, 717–723 CrossRef CAS.
- J.-C. Lin, Q.-X. Chen, X.-L. Xie, Z.-L. Zhuang, Y. Shi and Q.-S. Huang, J. Exp. Mar. Biol. Ecol., 2006, 339, 30–36 CrossRef CAS PubMed.
- Z.-X. Wang, H.-B. Wu, X.-C. Wang, H. Zhou and C.-L. Tsou, Biochem. J., 1992, 281, 285–290 CrossRef CAS.
- Y.-D. Park, Y. Yang, Q.-X. Chen, H.-N. Lin, Q. Liu and H.-M. Zhou, Biochem. Cell Biol., 2001, 79, 765–772 CrossRef CAS.
- J. Wu and Z. Wang, Biochem. J., 1998, 335, 181–189 CrossRef CAS.
- J.-J. Xie, Q.-X. Chen, Q. Wang, K.-K. Song and L. Qiu, Pestic. Biochem. Physiol., 2007, 87, 9–13 CrossRef CAS PubMed.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed.
- P. R. Palaniappan and V. Vijayasundaram, Infrared Phys. Technol., 2009, 52, 32–36 CAS.
- X. Zhao, F. Chen, W. Xue and L. Lee, Food Hydrocolloids, 2008, 22, 568–575 CrossRef CAS PubMed.
- C. R. Llerena-Suster, C. José, S. E. Collins, L. E. Briand and S. R. Morcelle, Process Biochem., 2012, 47, 47–56 CrossRef CAS PubMed.
- W. C. Johnson, Proteins: Struct., Funct., Bioinf., 1999, 35, 307–312 CrossRef CAS.
- N. Sreerama, S. Y. Venyaminov and R. W. Woody, Protein Sci., 1999, 8, 370–380 CrossRef CAS PubMed.
- A. Szabó, M. Kotormán, I. Laczkó and L. M. Simon, Process Biochem., 2009, 44, 199–204 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12068c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.