
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold-dotted hydroxyapatite nanoparticles as multifunctional platforms for medical applications
Catarina
Ferreira dos Santos
ab,
Pedro Sousa
Gomes
cd,
Maria Margarida
Almeida
e,
Marc-Georg
Willinger
f,
Ralf-Peter
Franke
g,
Maria Helena
Fernandes
cd and
Maria Elisabete
Costa
*e
aDepartment of Mechanical Engineering, Escola Superior de Tecnologia de Setúbal, Instituto Politécnico de Setúbal, Setúbal, Portugal
bICEMS, Instituto Superior Técnico, Technical University of Lisbon, Av. Rovisco Pais, Lisboa 1049 001, Portugal
cLaboratory for Bone Metabolism and Regeneration, Faculdade de Medicina Dentária, Universidade do Porto, Portugal
dMedInUP – Center for Drug Discovery and Innovative Medicines, University of Porto, Porto, Portugal
eDepartment of Materials and Ceramics Engineering, CICECO, Aveiro Institute of Materials, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: elisabete.costa@ua.pt
fMax Planck Gesell, Fritz Haber Inst, Dept Inorgan Chem, D-14195 Berlin, Germany
gCentral Institute for Biomedical Technology, Biomaterials Division, University of Ulm, Ulm, Germany
First published on 23rd July 2015
Abstract
A novel synthetic approach for coupling metallic gold nanoparticles (AuNPs) to the facets of hydroxyapatite crystalline nanoparticles (HapNPs) without an additional reducing agent was developed. Hexagonal prism-shaped hydroxyapatite nanoparticles (HapNPs) were first precipitated by a hydrothermal route assisted by citric acid, which left behind carboxylate species adsorbed on the HapNP facets. HapNP were then used as templates for gold precipitation. Upon exposure to gold ions, the carboxylate species on the HapNP prismatic facets were able to trigger the nucleation of metallic gold by reducing the gold ions in situ. The used conditions did not allow for the extensive growth of gold nuclei and hence gold dots with diameters not larger than 2.5 nm were formed on the HapNP facets. The obtained (Hap–AuNP) heteronanostructures combine Hap nanoprisms, a highly biocompatible material easily recognised by the human body, with metallic nanosized gold that imparts a plasmon resonance (SPR) effect to the synthesized nanostructures, which is of great potential for further exploration within the contexts of thermotherapy and imaging strategies. Human mesenchymal stem cells were exposed to a range of Hap–AuNP concentrations, but no cytotoxic effects from the heteronanostructures were observed. Furthermore, Hap–AuNP greatly enhanced Runx2 and ALP expressions. In addition, these results predict that the synthesized Hap–AuNP particles could hold potential interest for medical applications requiring nontoxic materials capable of bone-defect repair and furthermore exhibiting SPR properties, which can be potentially advantageous for therapeutic and diagnostic purposes.
1 Introduction
The unprecedented developments in materials synthesis and characterization techniques at the nanoscale level are encouraging audacious approaches in matter manipulation and assembly, which are being exploited for designing nanostructures with improved or new properties and functionalities compared with their bulk counterparts. Nanomedicine is one of the numerous fields wherein such cutting-edge nanotech advancements are fostering innovative strategies similar to that of cancer theranostics.1Gold nanoparticles (AuNP) are an emblematic example of biomedical nanostructures with a well-known surface plasmon resonance effect (SPR), the frequency of which is affected by nanoparticle (NP) size, shape, inter-distance and refractive index of surrounding NP media. This effect has been largely exploited for labeling and probing a wide variety of biomolecules that interfere with AuNP dispersed states and with the refractive index of the NP environment due to adsorption.2 Such applications demand a close control of AuNP morphological attributes (size and shape) and in particular, of AuNP dispersed conditions. These prerequisites have been covered by a large number of publications and hence various proposals for AuNP production, including synthetic methods, reagents, solvents, and stabilizers, are found in recent literature. Currently, AuNPs are also being explored as photothermal contrast agents for cancer imaging and therapy.3,4 Plasmonic AuNP strategically located in tumors are used to enhance near-infrared laser absorption and heat dissipation and thus produce a local temperature increase that disturbs cellular functions, resulting in cell death. In addition, the X-ray absorbing properties of AuNPs can be also used for biodistribution data acquisition enabling non-invasive tumor imaging and 3D reconstruction.3,5 Other treatment methods exploit spherical AuNP as sensitizing agents in radio frequency ablation and X-ray radiation cancer therapies.3,6–8
Hydroxyapatite (Hap) is an appealing inorganic biomaterial. Its chemical composition, crystalline structure and particle morphology if appropriately engineered can replicate the chemical and physical attributes of the inorganic components of human bone and teeth9,10 while offering enhanced adsorptive ability towards a considerable number of biomolecules and drugs.11,12 Addressing the combination of the exceptional properties of AuNPs and Hap and benefiting from the easy functionalization of AuNPs for specific protein binding, hybrid hydroxyapatite gold nanoparticles (Hap–AuNPs) structures with a portfolio of interesting properties for various bioapplications, such as immunosensing,13 bone-tissue regeneration12,14,15 and improvement of hemocompatibility,15,16 have been frequently reported. The methods found for the production and/or assembly of Hap–AuNPs structures are quite diverse.13–16 However, gold and Hap particles are often precipitated separately and then assembled to form the composite material.12,13,16,17 The Turkevich method18 or a variant of it is still seen as the method of choice for AuNP synthesis, whereas no preferred method could be identified for Hap synthesis because of the successful implementation of multiple techniques.19 Frequently, polymers (e.g. collagen and chitosan) are required as linkers or agents for the coupling of gold and Hap.14,15,17 One simple method for producing uniform Hap nanoparticles (HapNPs) is hydrothermal synthesis in the presence of an organic chelating ion or molecule20–23 used as a shape tailoring agent that allows the control of particle size and shape.24,25 According to recent reports, citrate ions could modulate prismatic HapNPs under specific hydrothermal synthetic conditions.23
The present study addresses AuNP nucleation and growth onto the surface of HapNPs for producing organized composite nanostructures. The ability of citric ions to play a dual role, i.e. as a HapNP tailoring agent and as an in situ reducing reagent of Au3+ leading to local Au0 nucleation and growth on HapNP facets is the engineering tool exploited herein. From an application perspective, widespread potential is anticipated, considering the benefits of obtaining accurate control over the size, shape and surface chemistry of AuNPs with the high biocompatibility of HapNP. Due to bone affinity, biomimetic features and osteointegrity, HapNP is particularly suited for bone-related applications.26,27 Thus, in addition to the interest in AuNP usage as an excellent contrast element28,29 depending on its size, the SPR tunability is envisaged with great usefulness for imparting simultaneous imaging and therapeutic abilities to Hap–AuNPs nanostructures, which makes them powerful tools for differentiated bone applications. Therefore, another point of interest is the discussion of the potential effects of HapNP assembled with AuNPs on the proliferation and differentiation of human mesenchymal stem cells due to their role in bone metabolism and regeneration, an issue that has not been covered so far.
2 Materials and methods
2.1 Hydroxyapatite synthesis
A previously reported hydrothermal precipitation method30 was followed for HapNP synthesis. The starting precursor solution was prepared in the following manner: a 0.6 M citric acid monohydrate (C6H8O7·H2O) (Riedel-deHaën, 99.5%) solution was mixed with a 0.2 M calcium nitrate Ca(NO3)24H2O (Riedel-deHaën, 99%) solution and a 0.2 M ammonium hydrogen phosphate (NH4)2HPO4 (Merck, 99%) solution was then added. The pH of the solution was adjusted to 8.1 with small additions of a 25% ammonia solution (Riedel-deHaën). The final solution was poured into a 100 ml Teflon vessel, filling up 50% of the total vessel volume. The Teflon vessel was then sealed in a stained-steel autoclave and subsequently transferred into an oven at 180 °C wherein it was held under autogenous pressure for 24 hours. The autoclave was then quenched in water at room temperature. Finally, the precipitated particles were collected and filtered using a glass Millipore filter vessel and then dried in a desiccator.2.2 Gold precipitation
For the precipitation of gold, 86.5 μl of a hydrogen tetrachloroaurate (HAuCl4) solution was added to 500 ml of deionized water in order to obtain a 0.25 × 10−3 M gold solution with pH of ∼3.25 ml of the gold solution was then heated at ∼100 °C for 3 min and 50 mg of HapNP was subsequently added, which was synthesized as described in Section 2.1. The resulting suspension was maintained under boiling conditions for a period of 5 min. The overall procedure was repeated for covering various reaction times, i.e. boiling times of 10 min and 20 min. The particles (Hap–AuNP) were then repeatedly washed with deionized water during filtration with a glass Millipore filter vessel and then dried in a desiccator.2.3 Particle characterization
The morphology of all particles obtained by precipitation was evaluated through electron transmission microscopy (TEM) using a Hitachi H-9000-NA microscope operated at 200 kV and high-resolution electron transmission microscopy (HRTEM) images were obtained on a JEOL 2200 FS microscope operating at 200 kV. For TEM analysis, the particles were dispersed with 2-propanol and scattered over copper grids coated with a formvar film, and TEM images were used to determine AuNP sizes. More than 100 particles were randomly counted to determine the particle size distribution using Image-J software. Particle crystalline phases were identified by powder X-ray diffraction (XRD) analysis (Rigaku PMG-VH with CuKα radiation = 1.5405 Å). Fourier transform infrared spectroscopy (FTIR) was performed to identify the functional groups using a Mattson Galaxy 3020 spectrophotometer. The test samples were prepared by mixing ∼2 mg of the precipitated particles with ∼300 mg of spectroscopic-grade KBr (Merck), followed by shaping the mixture into a disk. The infrared spectra were acquired in transmittance mode in the region of 4000–400 cm−1 with a resolution of 4 cm−1. The UV-vis absorption data were collected on a Shimadzu MPC-3100 PC series spectrophotometer using an auto scan between 200 and 700 nm. The specific surface area (SSA) of the obtained particles was determined through N2 adsorption using a Micromeritics Gemini 2370 V5 Series. The samples were thoroughly degassed for 24 h at 120 °C before nitrogen adsorption. The SSA was determined based on the multipoint Brunauer–Emmett–Teller isotherm (BET).202.4 Interaction of Hap–AuNPs with human mesenchymal stem cells
Human mesenchymal stem cells–bone marrow-derived (HMSC-bm, Innoprot), according to the supplier, were found to stain positive for CD44 and CD90, which are characteristic markers of the population phenotype. Third subculture cells were used to evaluate the cell response to the nanoparticles.HMSC, 104 cells per cm2, was cultured in Minimum Essential Medium Eagle, alpha modification (α-MEM, Sigma-Aldrich) containing 10% fetal bovine serum (FBS, Sigma-Aldrich), 50 μg ml−1 ascorbic acid, penicillin (10 units per ml)/streptomycin (2.5 μg ml−1; P/S solution, Sciencell) and 2.5 μg ml−1 of Fungizone. After 24 h, the medium was removed and a completely fresh culture medium containing sterilized HapNP or Hap–AuNPs—1, 10, 100 and 500 μg ml−1—was added to the adherent cells. Cultures were further incubated for 1, 3 and 7 days without any medium change. Cultures without nanoparticles were used as controls. Incubation was carried out under a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Cultures were characterized in the following manner at days 1, 3 and 7.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in PBS (20 min). Cells were counterstained with propidium iodide (Sigma Aldrich), 10 mg ml−1 in PBS (5 min). Stained samples were mounted in Vectashield® (Vector laboratories) and examined in a Leica SP5 AOBS (Leica Microsystems®) microscope.
:100 in PBS (20 min). Cells were counterstained with propidium iodide (Sigma Aldrich), 10 mg ml−1 in PBS (5 min). Stained samples were mounted in Vectashield® (Vector laboratories) and examined in a Leica SP5 AOBS (Leica Microsystems®) microscope.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GAPDH | CAGGACCAGGTTCACCAACAAGT | GTGGCAGTGATGGCATGGACTGT |
| Runx2 | CAGTTCCCAAGCATTTCATCC | TCAATATGGTCGCCAAACAG |
| COL1 | TCCGGCTCCTGCTCCTCTTA | ACCAGCAGGACCAGCATCTC |
| ALP | ACGTGGCTAAGAATGTCATC | CTGGTAGGCGATGTCCTTA |
| OC | CACTCCTCGCCCTATTG | CCCACAGATTCCTCTTCT |
| OPG | AAGGAGCTGCAGTAGGTCAA | CTGCTCGAAGGTGAGGTTAG |
3 Results and discussion
3.1 Synthesis of functionalized hydroxyapatite nanosized particles (HapNP)
Literature reports on hydroxyapatite synthetic methods are plentiful. The choice of a particular synthetic methodology is normally dictated by the particle morphology and/or crystallinity criteria. In the present case, a previously reported hydrothermal precipitation method23 was selected to obtain crystalline HapNP with nanometric dimensions and well-defined facets easily imaged using electron microscopy. In addition, other issues, including the surface chemistry of the precipitated particles, were taken into account.Fig. 1(a) shows an image of HapNP as synthesized by the hydrothermal procedure. As observed, the particles display a light-yellowish colour. The crystalline phase composition of these particles is revealed by the XRD pattern presented in Fig. 2 (curve (a)), the diffraction peaks of which can be assigned to hydroxyapatite (Hap), according to the JCPDS no. 09-0432. Fig. 3(a) shows the FT-IR spectrum of the precipitated HapNP in which the characteristic peaks of hydroxyapatite are detected.33–35 Furthermore, several features attributed to the organic groups are also identified in a range from 1680 cm−1 to 1350 cm−1 (Fig. 3(b)). The band at 1384 cm−1 may be ascribed to CH2 scissoring, and carboxylate groups differently coordinated to the Hap surface under monodentate or bidentate configurations6,30 may account for the bands at 1402, 1458, 1577 and 1669 cm−1. Such adsorbed carboxylate species [R(COO−)x]ads can be interpreted as the original citrate ion fingerprints that might have decomposed under the temperature and pressure regime used for Hap synthesis, in line with the reports on citric acid thermal degradation.36–39
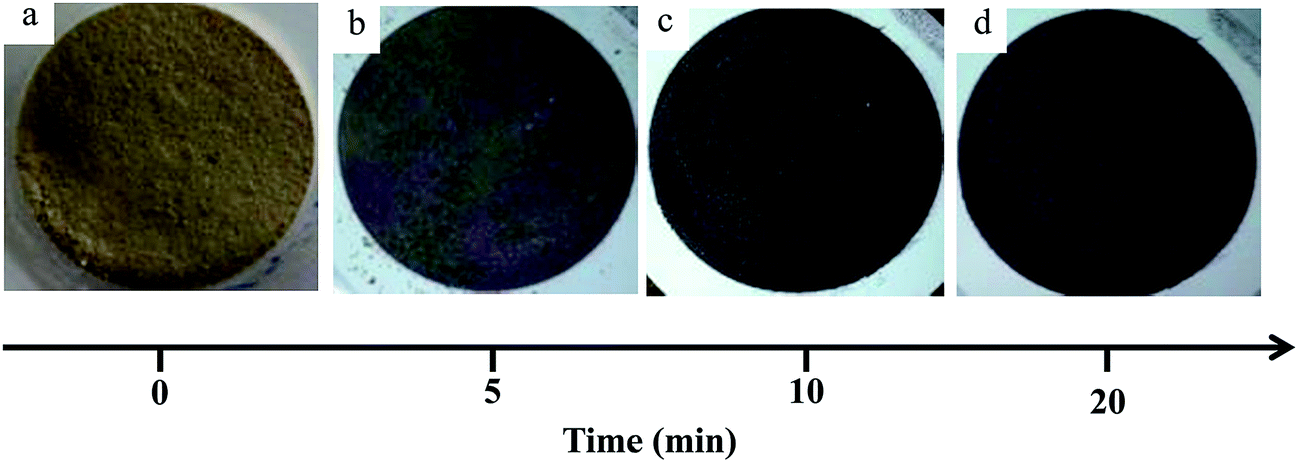 | ||
| Fig. 1 Optical images of hydroxyapatite nanoparticles: (a) as synthesized at 180 °C (HapNPs) and after submission to gold precipitation (Hap–AuNPs) during (b) 5 min, (c) 10 min and (d) 20 min. The particle colour evolution denotes the presence of metallic gold. | ||
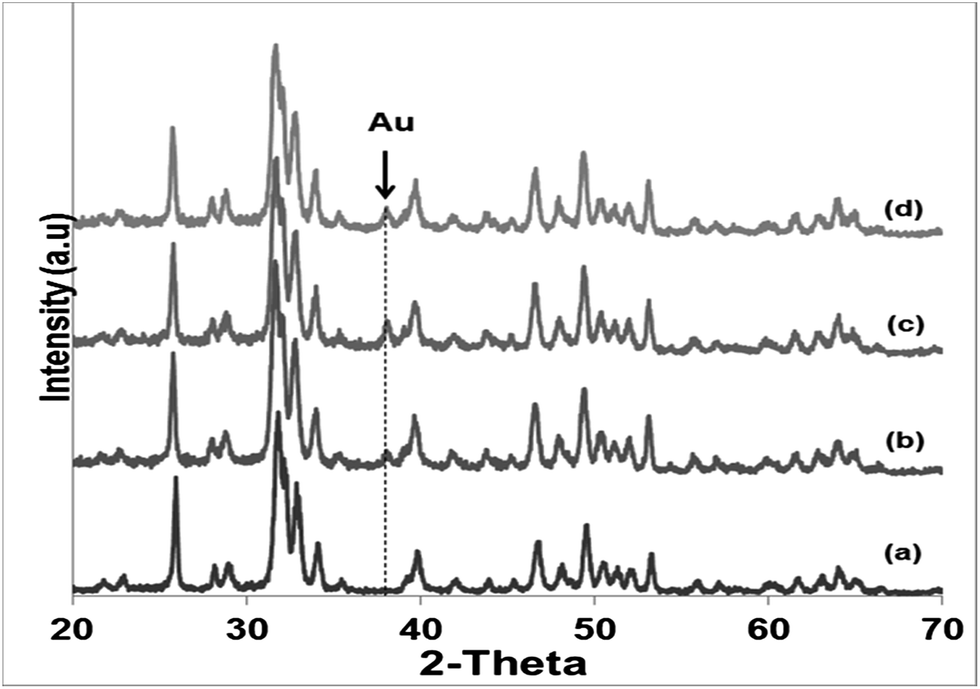 | ||
| Fig. 2 X-ray diffraction patterns of hydroxyapatite nanoparticles: (a) as produced at 180 °C (HapNPs) and after submission to gold precipitation (Hap–AuNPs) during (b) 5 min, (c) 10 min and (d) 20 min. The peak detected at 2θ ∼ 38° is assigned to metallic gold. | ||
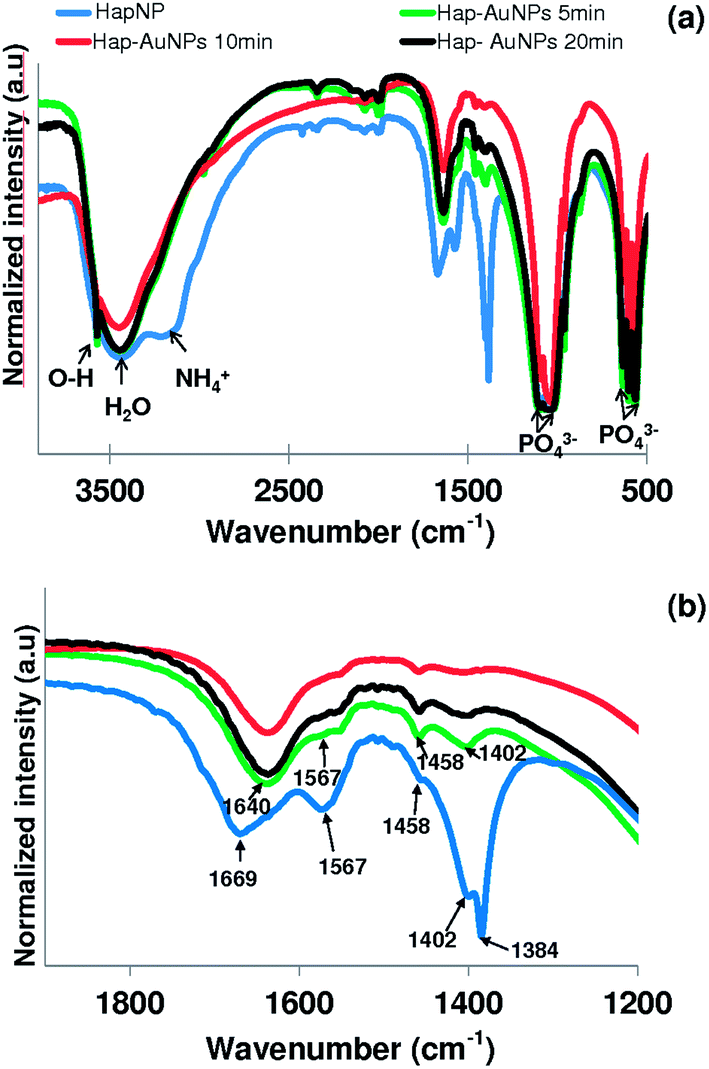 | ||
Fig. 3 FTIR spectra of (a) HapNPs synthesized at 180 °C ( ) and its evolution with metallic gold precipitation after 5 min ( ) and its evolution with metallic gold precipitation after 5 min ( ), 10 min ( ), 10 min ( ) and 20 min ( ) and 20 min ( ); (b) the 1200–1900 cm−1 spectral region is amplified. ); (b) the 1200–1900 cm−1 spectral region is amplified. | ||
The particle morphology assessed by TEM and shown in Fig. 4(a) and (b) can be well described by a hexagonal prismatic shape with a width (w) of between 20 and 30 nm and a length (l) ranging from 50 nm to 100 nm. These nanoscale dimensions of the prismatic hydroxyapatite nanoparticles (HapNP) account for a large specific surface area of 55 m2 g−1 and an aspect ratio (l/w) larger than 1.7. Fig. 4(b) shows the HRTEM image of a HapNPs wherein well-defined lattice fringe patterns are observed. The distance between adjacent lattice fringes corresponds to an interplanar distance of 0.65 nm that can be indexed to the d-spacing values of the hydroxyapatite crystal (010) planes, which are parallel to the c axis. The applied citrate-mediated hydrothermal synthesis thus favored preferential HapNP growth along the c-axis, which resulted in elongated particle morphology.
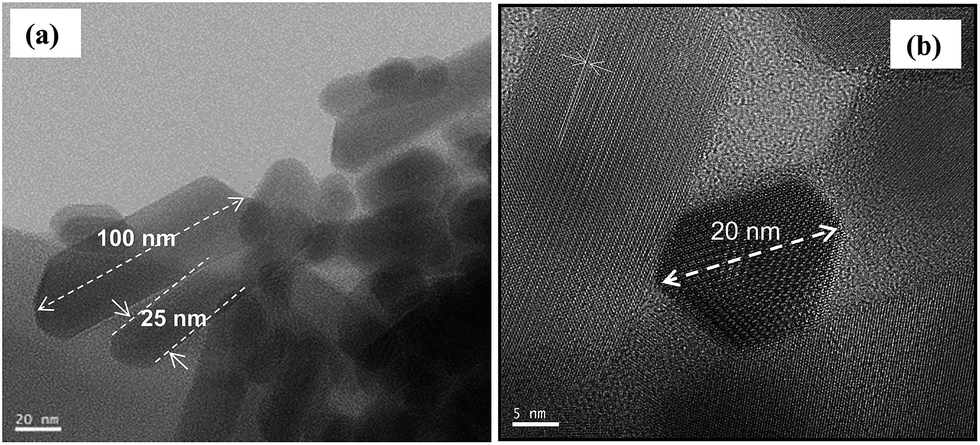 | ||
| Fig. 4 (a) TEM images showing the elongated prismatic shape of HapNPs. (b) The hexagonal base of prismatic particles is confirmed on a particle top view. | ||
The interaction of citric acid with the HapNP surface and its effects on HapNP growth have been frequently addressed, from both experimental and theoretical perspectives. Calcium phosphate precursor solutions containing citric acid have been reported to favor the precipitation of HapNP with elongated shapes under varied conditions of temperature and pressure.23 According to computer simulation studies,33,40 a strong binding of citric acid or of citrate ion to Hap lateral prismatic facets may slow their growth rates and as a result, enable their dominant expressions on the final particle morphology. Such theoretical predictions might explain the current results assuming that citrate ions at an early stage of the hydrothermal processing and citrate-derived moieties at a later stage of the precipitation perform as lateral growth inhibitors, hence contributing to particle shapes. An accurate illustration of citrate's role under the temperature and pressure regime of this study requires a complete follow-up of the interplay among Hap nucleation, growth and citrate degradation progress, which is now an ongoing study.
3.2 Templating gold precipitation with HapNP
HapNP synthesized by a hydrothermal technique were used as substrates for AuNP growth. The effects of gold precipitation time were examined to assess its impact on the gold particle size and size distribution.Fig. 5(a) and (b) shows the HRTEM images of the Hap–AuNPs particles collected after various precipitation times. It is clearly observed that the prismatic HapNP previously imaged in Fig. 4(a) are now uniformly spotted with nanosized dark dots, the dimensions of which vary approximately from 1.5 to 2.5 nm as the synthesis time increases from 5 min (Fig. 5(a)) to 20 min (Fig. 5(b)). These are nanosized dots of metallic gold that were precipitated on HapNP facets, accounting for the HapNP colour variation previously described. It was also found that Hap–AuNPs after 20 min of reaction time apparently have a lower density of larger AuNPs dots on their surfaces as documented by the particle-size distribution curves (Fig. 5(c)). This may be explained by an Ostwald ripening mechanism favouring the growth of larger AuNPs at the expense of the smaller.19 Although the dots become larger, they apparently preserve the spherical shape, which is known to be the lowest-energy shape, often observed among metal nanoparticles obtained from the reaction of metal salts with reducing agents.42 The present strategy proved to be highly reproducible, consistently coupling uniform spherical AuNPs to HapNPs.
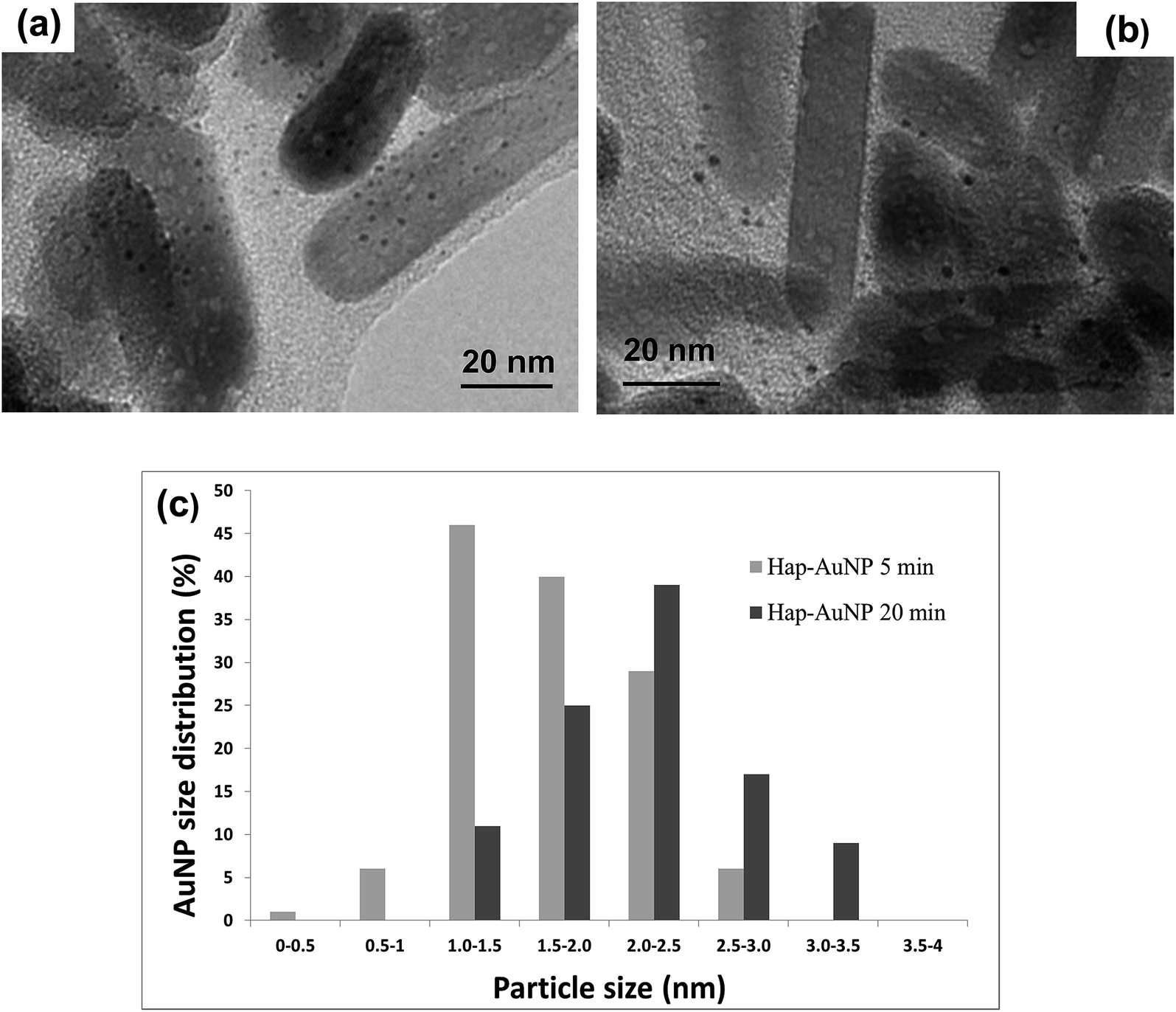 | ||
| Fig. 5 HRTEM images of Hap–AuNPs collected after two different precipitation times: (a) 5 min and (b) 20 min. The particles are spotted with nanosized dark dots of metallic gold (AuNP), which were precipitated on HapNP facets. (c) AuNP size distribution precipitated after 5 min and 20 min. | ||
Fig. 6 shows the UV-vis spectra of as-prepared HapNP (a) and of Hap–AuNPs after 5 min (b) and 20 min (c) of synthesis. No UV-vis band is detected in the range of 300–800 nm for as-prepared HapNP, which confirms previous reports.41 In contrast, a characteristic SPR band of AuNPs is clearly observed around 550 nm in the Hap–AuNPs spectra. The observed SPR accounts for the strong colour changes observed on the particles29 and confirms the formation of spherical AuNP by this synthetic method, which is in good agreement with the TEM results. Moreover, it was observed that the SPR band is similar for the two selected precipitation times, which may be related to the small variation in particle size, which is insignificant for detection by using UV-vis in powders. The SPR band shift from the well-known value of 520 nm reported for isotropic spherical gold particles (<20 nm) dispersed in water43,44 to the observed value herein of ∼550 nm may reflect the influence of the substrate (Hap) refractive index (1.64), which is larger than that of water (1.33), to which the surrounding medium is generally referred.45 A similar shifting effect was also reported for gold nanoparticles of ∼5.5 nm deposited over SiO2/Si substrates.46
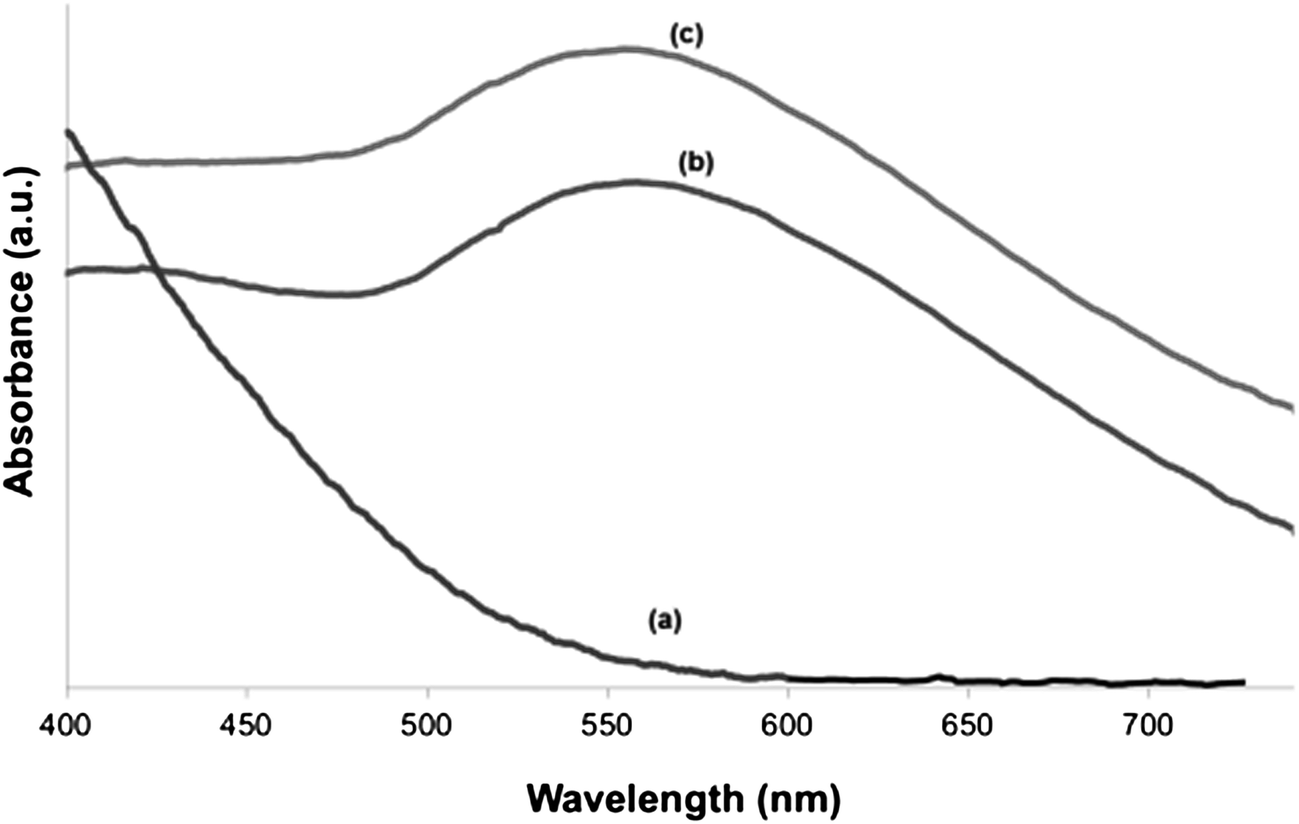 | ||
| Fig. 6 UV-vis spectra of (a) HapNPs synthesized at 180 °C and after reacting with the gold solution (Hap–AuNPs) during (b) 5 min and (c) 20 min. The characteristic surface plasmon resonance band of gold nanoparticles is clearly observed at ∼552 nm for the Hap–AuNPs. | ||
As referred by Kumar et al.,48 gold precipitation in a bulk solution by the Turkevich method can be systematized according to the following steps. An initial step comprises the oxidation of citrate (CH2COO−)2C(OH)COO− to an acetone dicarboxylic acid (CH2COO−)2CO accompanied by the reduction of auric salt (AuCl3) to aurous salt (AuCl). The subsequent step requires the disproportionation of AuCl into gold atoms (Au0) and AuCl3, as schematized by the equation 3AuCl → 2Au0 + AuCl3. This last step requires various aurous chloride molecules to be in a nearby condition, which is facilitated by an acetone dicarboxylic species that tethers molecular aurous chloride.
In this study, gold precipitation took place on the HapNP surface, most likely on the lateral prismatic faces as confirmed by TEM images. When HapNPs are added to the reagent HAuCl4 aqueous solution (pH of ∼3), HapNPs acquire a neutral or weakly positive zeta potential23 that favours electrostatic attachment of negatively charged auric complexes, i.e. [AuCl3OH]− and/or [AuCl2OH2]−, which are abundant species in an HAuCl4 solution at pH 3–4.49,50 It is herein proposed that [R(COO−)x]ads groups undergo oxidation under the thermal conditions of the precipitation process (T = 100 °C), thus enabling the reduction of the auric complexes on the HapNP surface to aurous species and mimicking the role of acetone dicarboxylic acid reported by Kumar et al.48 Furthermore, the vicinity of the resulting aurous species will present an advantageous condition for the disproportionation reaction to proceed towards Au0 nucleation.
Although previous reports have shown that gold precipitation may take place on pristine oxide surfaces wherein O− or OH− are the protagonist surface species interacting with solution gold complexes,49–51 long precipitation times are thus required (≥1 h) compared with the few minutes reported in this study. The applied HapNPs thus behaved as a substrate that prompted the heterogeneous nucleation of AuNPs without the addition of any reducing agent such as NaBH4, one of the most commonly used reagents for gold ion reduction.44 Another method for the deposition of gold on the surface of Hap nanoparticles was reported by J.-D. Wang et al.52 By using urea as a precipitating agent, a gold precursor (Au(OH)3) was precipitated, which was later converted to metallic gold upon heat treatment at 350 °C. Therefore, compared with that synthesis route, the present method offers a straightforward way to produce metallic gold, exempting any precipitating agent or intermediate calcination step. The carboxylic species attached to the HapNP surface are thus a strategic tool for in situ gold reduction and nucleation hence leading to AuNPs uniformly distributed on HapNP facets, without the need of any additional surfactant for stabilization.19 Being attached to HapNP surfaces, AuNPs are not prone to aggregation, thereby eliminating potential drawbacks associated with aggregates.
Recent reports examined the multiple functionalities attributed to AuNP, several of which in a concerted manner may convey performance synergy in the field of therapeutics.53,54 In the present case, the coupling of AuNPs to HapNPs imparts an SPR effect to the resulting composite nanostructure, which could be further exploited for thermotherapy: AuNPs are envisaged to empower HapNPs with hyperthermic capability for treating near-surface tumours or skin-type cancers wherein laser requirements are less strict as compared to tumours located deep within bodily tissue for which a laser light in the near-infrared (NIR) region of the biological water window (650–900 nm) would be recommended.29 Moreover, HapNPs can also behave as drug delivery systems (DDS)55 due to their ability in the adsorption and release of therapeutic drugs.55 The endowment of a thermal response afforded by AuNPs to such DDS could open the possibility of customizing a HapNP-based DDS to a target of interest. It was reported that the addition of a thermo-responsive coating to a DDS would allow for the use of induced thermal stimulus to trigger drug delivery only at the local area of interest,56 thus confining the drug effects to the unhealthy tissue while minimizing drug losses before the diseased cells are reached. In addition to thermotherapeutic benefits, AuNPs may also provide imaging capabilities as demonstrated in a recent study wherein the ability of gold nanoparticles to perform as a CT imaging contrast agent for tumour imaging was reported.54 Therefore, the hydroxyapatite-based nanostructures (Hap–AuNPs) engineered in the present study offer promising characteristics for thermotherapeutic and imaging functionalities. The exploration of such attributes will warrant significant interest because it may lead to the expansion of hydroxyapatite applications beyond its well-known medical use in hard-tissue regeneration.
3.3 Interaction of Hap–AuNPs with human mesenchymal stem cells
As previously mentioned, because of the similarity of Hap to the mineral phase of bone tissue and thus its natural bone affinity and biocompatibility, a range of bone-tissue-related applications are envisaged for HapNPs.26,27 In a previous study, HapNPs similar to those reported herein proved to be non-toxic to MG63 osteoblastic cells in terms of cell viability/proliferation, F-actin cytoskeleton organization and apoptosis rate. In addition, they increased the expression of ALP and BMP-2.23 In order to assess the potential of the synthesized Hap–AuNPs for bone-related applications, the particles obtained after 5 min of reaction time were selected to be cultured with human bone marrow – derived mesenchymal stem cells – due to their potential in bone regenerative approaches. HMSC were cultured under standard culture conditions, with the medium supplemented by ascorbic acid, because of its role in the synthesis of the bone collagenous extracellular matrix57 but in the absence of any osteogenic inducer. HMSC cell response to the NPs was assessed for cell viability/proliferation and osteoblastic-related markers.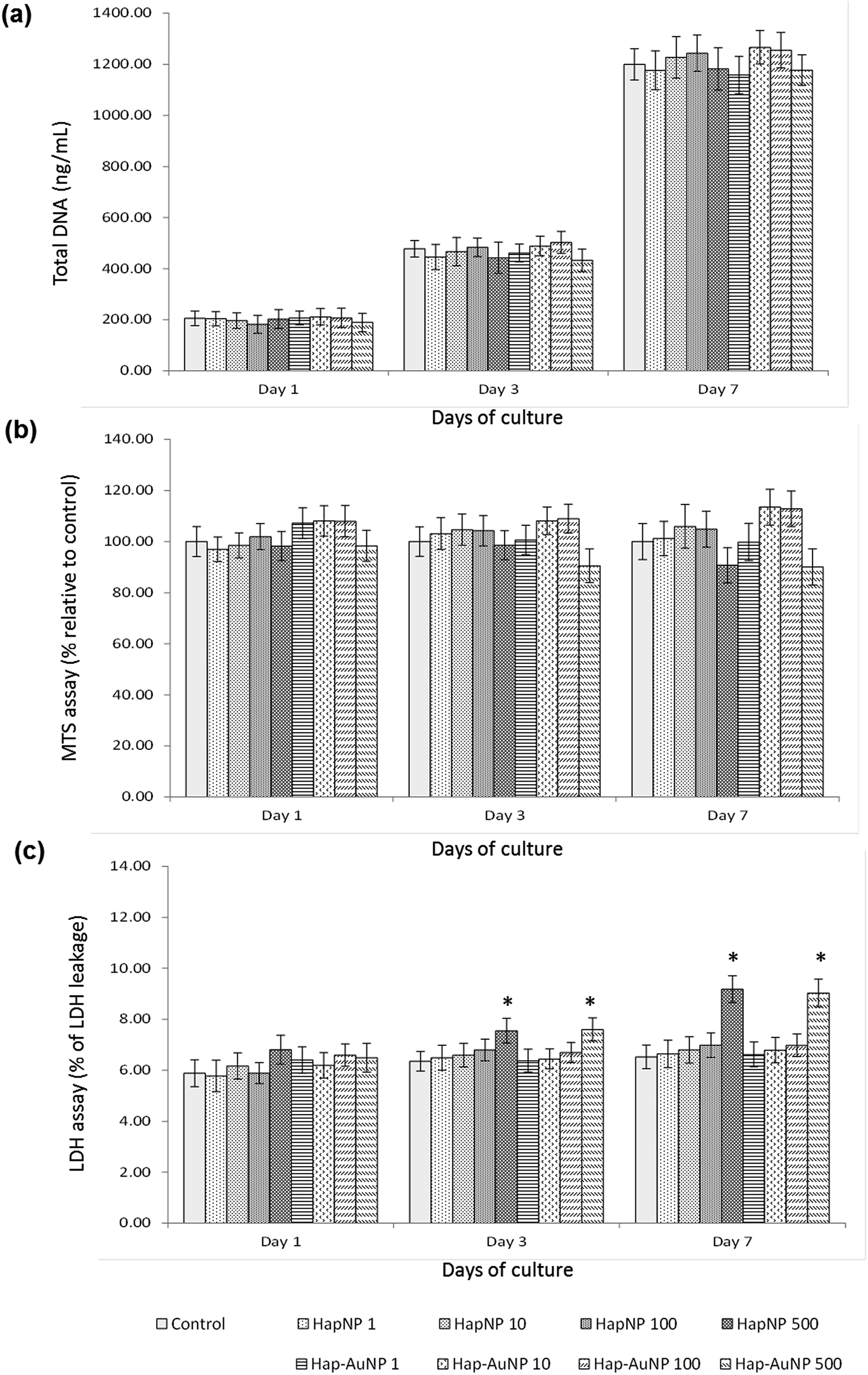 | ||
| Fig. 7 (a) DNA content, (b) MTS reduction and (c) LDH leakage of HMSC cultured for 1, 3 and 7 days in the presence of HapNPs and Hap–AuNPs, 1–500 μg ml−1. NPs did not interfere with the cellular proliferation and metabolic activity in the range 1–100 μg ml−1, but exposure to 500 μg ml−1 caused a slight decrease in cellular viability. *Significantly different from control (absence of NPs). | ||
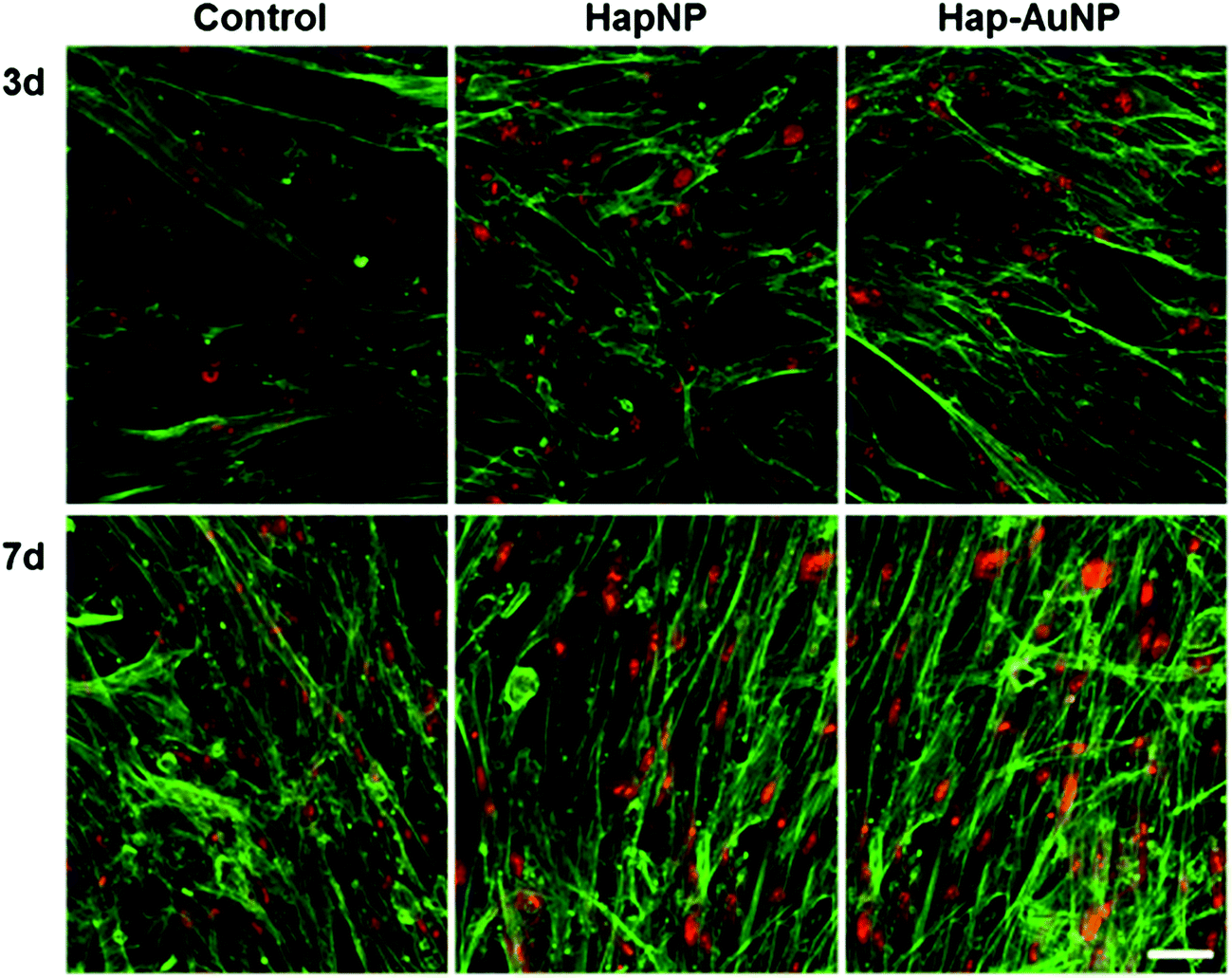 | ||
| Fig. 8 Representative confocal laser scanning microscopy images of HMSC cultured for 3 and 7 days in the presence of HapNPs and Hap–AuNPs, 100 μg ml−1. Cultures were stained for the cytoskeleton (green) and nucleus (red). Cellular morphology and organization of the cell layer were similar in control and NP-exposed cultures. Bar: 50 μm. | ||
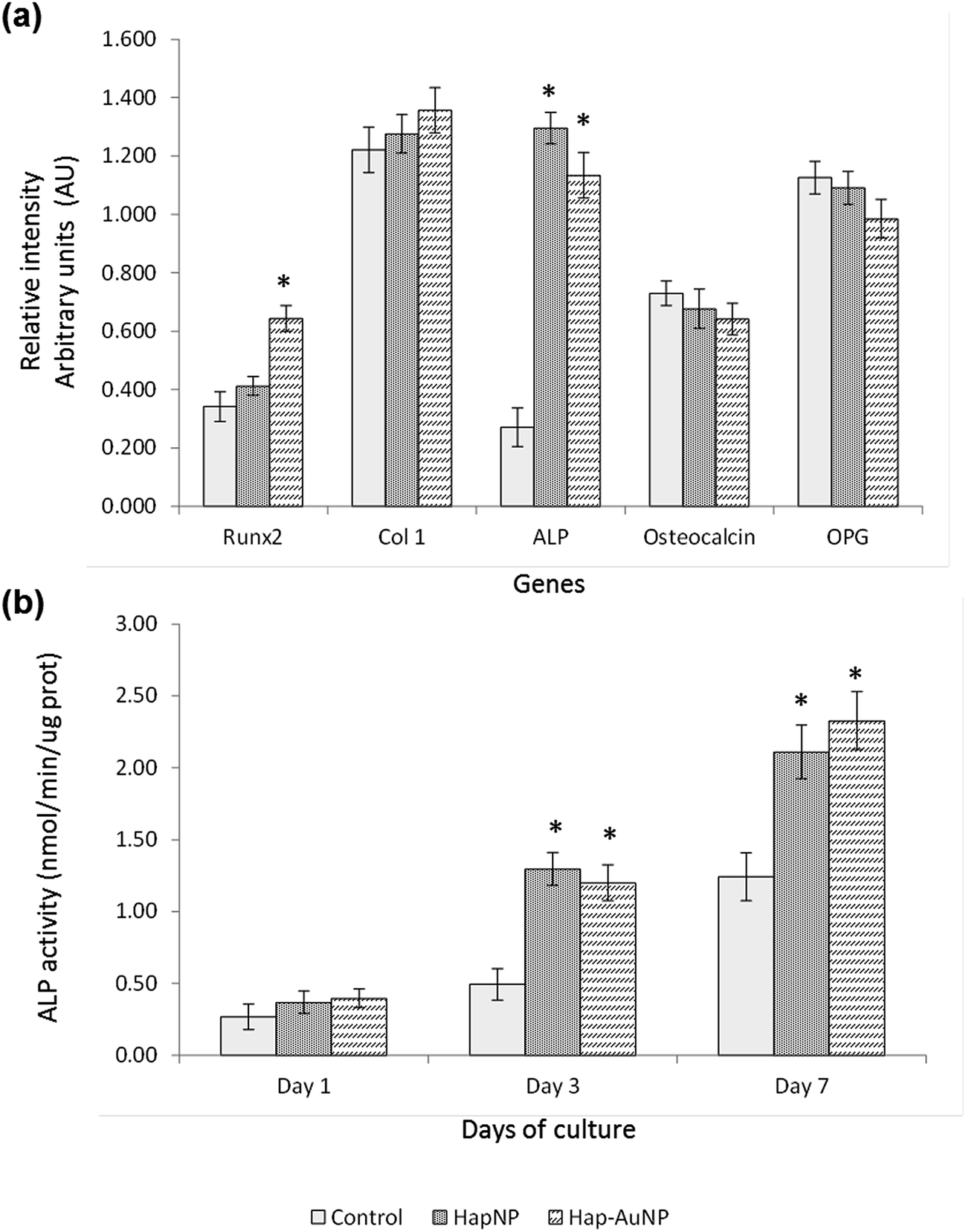 | ||
| Fig. 9 (a) Gene expression and (b) ALP activity of HMSC cultured in the presence of HapNPs and Hap–AuNPs, 100 μg ml−1; (a) cultures with 3 days; (b) cultures with 1, 3 and 7 days. *Significantly different from control (absence of NPs). Hap–AuNPs caused a significant increase in the expression of Runx2, and both NPs increased ALP expression and activity. | ||
Literature reports on the interaction of osteoblastic lineage cells with Hap nanoparticles show a great variability of results, which is expected considering the wide-ranging versatility and diversity of the physicochemical properties of the tested particles, cell system and experimental protocols.23,59–63 However, regarding the present HapNP, the observed cell response is in line with previous reports showing low cytotoxicity of Hap nanoparticles towards osteoblastic cells, in addition to their ability to modulate cell proliferation and differentiation events. Upon exposure to HapNP, increased proliferation was found in bone marrow-derived mesenchymal stem cells,63 and enhanced osteoblastic differentiation features were also observed for these cells,59,63 in addition to the osteoblastic cell lines hFOB 1.19 (ref. 62) and MG63.23 On the other hand, cytotoxicity/biocompatibility studies involving AuNPs coupled with hydroxyapatite are rare or even non-existent. In regard to AuNPs, the osteoblastic cell response also showed variable results. In the murine preosteoblastic cell line MC3T3-E1, AuNPs did not affect cellular viability;64 in another study, they promoted their viability and osteogenic differentiation, depending on the particle size.65 In mesenchymal stem cells, AuNPs promoted viability and osteogenic differentiation,66 but they were also reported to be toxic to these stem cells, depending on the particle size.67 AuNp aggregation states also influence cellular uptake and cytotoxicity as reported for various cell types,68,69 although not following a simple rule of thumb as aggregated AuNP may result in cytotoxicity or favour cell growth depending on the aggregate size, which in turn conditions its internalization.68 The conflicting results could also arise from the variability of the applied toxicity assays, cellular systems, and chemical/physical properties of the nanoparticles; moreover, the dosing parameters and exposure times to AuNPs vary, which make comparisons difficult. In the present study, the synthesized Hap–AuNPs showed low cytotoxicity, and this may reflect the use of a system in which small-sized AuNPs are fixed to a HapNP template and thus inhibited from undergoing agglomeration. Hap–AuNPs also exhibited the ability to enhance early phenotype markers for osteogenic differentiation in HMSC, i.e. ALP expression and activity. Furthermore, the considerable increase in the expression of Runx2 is significant, pointing to a higher commitment of mesenchymal stem cells toward the osteogenic lineage in the presence of these particles. In this regard, it has been reported that AuNPs are able to activate the p38 MAPK signalling pathway in the MSCs, which leads to the up-regulation of the osteogenic master transcription factor Runx2, driving MSCs to differentiate toward osteoblast cells.66
The potential of AuNPs for use in numerous different biological applications has led to strong interest in their cellular and tissue compatibility. In this regard, the response of the synthesized Hap–AuNPs towards mesenchymal stem cells seems to substantiate interesting features in bone-related applications from imaging and drug delivery to regenerative approaches. However, their ability to affect the fate of mesenchymal stem cells, i.e. by the enhancement of osteogenic differentiation pathways, should also be carefully considered in bone-related and other biomedical applications.
4 Conclusions
Gold nanoparticles (AuNPs) were successfully synthesized on the surface of hydroxyapatite nanoparticles (HapNP). The presence of organic species containing carboxylate groups on HapNP surfaces obtained by the synthetic processes of the HapNPs were a key condition for enabling in situ ionic gold reduction to metal gold, which resulted in spherical AuNPs with a diameter of few nanometers. The presence of nanometric gold on the resulting composite particle (Hap–AuNPs) imparts an SPR effect to the final particles, which may be further explored for imaging and therapeutic purposes. Hap–AuNP interaction with mesenchymal stem cells converged to confirm the nanoparticles cytocompatibility and also their enhancing osteoblastic differentiation properties. In addition, this profile substantiates Hap–AuNPs as promising materials for bone-related applications.Acknowledgements
This study was financed by the FEDER, via ‘‘Programa Operacional Factores de Competitividade – COMPETE′′, FCT – ‘‘Fundação para a Ciência e a Tecnologia’’, PEst-C/CTM/LA0011/2011, and FMDUP. The authors are grateful to the Portuguese Electronic Microscopy Network for support through FCT funding. CLSM observation was performed at Advanced Light Microscopy, IBMC, University of Porto (IBMC.INEB) under the direction of Dr Paula Sampaio.References
- Y. Xia, Nat. Mater., 2008, 7, 758–760 CrossRef CAS PubMed.
- Y. Zhang, Y. Guo, Y. Xianyu, W. Chen, Y. Zhao and X. Jiang, Adv. Mater., 2013, 25(8), 3802–3819 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41(7), 2740–2779 RSC.
- G. von Maltzahn, J.-H. Park, K. Y. Lin, N. Singh, C. Schwöppe, R. Mesters, W. E. Berdel, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Nat. Mater., 2011, 10, 545–552 CrossRef CAS PubMed.
- G. von Maltzahn, J.-H. Park, A. Agrawal, N. K. Bandaru, S. K. Das, M. J. Sailor and S. N. Bhatia, Cancer Res., 2009, 69(9), 3892–3900 CrossRef CAS PubMed.
- K. Kim, D. Dean, A. Lu, A. G. Mikos and J. P. Fisher, Acta Biomater., 2011, 7(3), 1249–1264 CrossRef CAS PubMed.
- M. Patel, K. J. Patel, J. F. Caccamese, D. P. Coletti, J. J. Sauk and J. P. Fisher, J. Biomed. Mater. Res., Part A, 2010, 94(2), 408–418 Search PubMed.
- S. Yang, D. Wei, D. Wang, M. Phimphilai, P. H. Krebsbach and R. T. Franceschi, J. Bone Miner. Res., 2003, 18(4), 705–715 CrossRef CAS PubMed.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58(3–5), 77–116 CrossRef.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12(18), 4329–4335 CrossRef.
- S. Aryal, R. K. C. Bahadur, S. R. Bhattarai, P. Prabu and H. Y. Kim, J. Mater. Chem., 2006, 16, 4642–4648 RSC.
- Y. Ding, J. Liu, H. Wang, G. Shen and R. Yu, Biomaterials, 2007, 28(12), 2147–2154 CrossRef CAS PubMed.
- T. P. Sastry, J. Sundaraseelan, K. Swarnalatha, S. S. L. Sobhana, M. U. Makheswari, S. Sekar and A. B. Mandal, Nanotechnology, 2008, 19, 245604 CrossRef CAS PubMed.
- Y. Gupta, G. N. Mathur and S. Verma, Bioorg. Med. Chem. Lett., 2006, 16(2), 363–366 CrossRef CAS PubMed.
- S. Arumugan, A. M. Rajam, N. Natarajan, U. Rao, C. Rose and T. P. Sastry, J. Biomed. Nanotechnol., 2006, 2, 46–52 CrossRef.
- K. C. R. Bahadur, S. Aryal, N. Bhattarai and H. Y. Kim, Scr. Mater., 2006, 54(12), 2029–2034 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. B. Hannon, S. Kodambaka, F. M. Ross and R. M. Tromp, Nature, 2006, 440, 69–71 CrossRef CAS PubMed.
- Y. Wang, S. Zhang, K. Wei, N. Zhao, J. Chen and X. Wang, Mater. Lett., 2006, 60(12), 1484–1487 CrossRef CAS.
- C. R. Kothapalli, M. Wei, R. Z. Legeros and M. T. Shaw, J. Mater. Sci.: Mater. Med., 2005, 16, 441–446 CrossRef CAS PubMed.
- H. Arce, M. L. Montero, A. Sáenz and V. M. Castaño, Polyhedron, 2004, 23(11), 1897–1901 CrossRef CAS.
- C. Santos, P. S. Gomes, J. A. Duarte, R. P. Franke, M. M. Almeida, M. E. V. Costa and M. H. Fernandes, J. R. Soc., Interface, 2012, 9, 3397–3410 CrossRef CAS PubMed.
- M. A. Martins, C. Santos, M. M. Almeida and M. E. V. Costa, J. Colloid Interface Sci., 2008, 318, 210–216 CrossRef CAS PubMed.
- S. P. Garcia and S. Semancik, Chem. Mater., 2007, 19(16), 4016–4022 CrossRef CAS.
- K. Fox, P. A. Tran and N. Tran, ChemPhysChem, 2012, 13(10), 2495–2506 CrossRef CAS PubMed.
- H. Zhou and J. Lee, Acta Biomater., 2011, 7, 2769–2781 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103(40), 8410–8426 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Plasmonics, 2007, 2(3), 107–118 CrossRef CAS.
- P. Schneider, Appl. Catal., A, 1995, 129, 157–165 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- T. Decker and M.-L. Lohmann-Matthes, J. Immunol. Methods, 1988, 115, 61–69 CrossRef CAS PubMed.
- N. H. D. Leeuw and J. A. L. Rabone, CrystEngComm, 2007, 9, 1178–1186 RSC.
- B. A. J. Noordover, R. Duchateau, R. A. T. M. van Benthem, W. Ming and C. E. Koning, Biomacromolecules, 2007, 8, 3860–3870 CrossRef CAS PubMed.
- D. Jiang and J. Zhang, Curr. Appl. Phys., 2009, 9, S252–S256 CrossRef.
- S. Bednarz, M. Lukasiewicz, W. Mazela, M. Pajda and W. Kasprzyk, J. Appl. Polym. Sci., 2011, 119, 3511–3520 CrossRef CAS.
- D. Wyrzykowski, E. Hebanowska, G. Nowak-Wiczk, M. Makowski and L. Chmurzyński, J. Therm. Anal. Calorim., 2011, 104, 731–735 CrossRef CAS.
- T. B. Mostafa, H. F. Naguib, M. W. Sabaa and S. M. Mokhtar, Polym. Int., 2005, 54, 221–225 CrossRef CAS.
- G. D. Cody, N. Z. Boctor, R. M. Hazen, J. A. Brandes, H. J. Morowitz and H. S. Yoder Jr, Geochim. Cosmochim. Acta, 2001, 65(20), 3557–3576 CrossRef CAS.
- S. R. Qiu, A. Wierzbicki, C. A. Orme, A. M. Cody, J. R. Hoyer, G. H. Nancollas, S. Zepeda and J. J. de Yoreo, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1811–1815 CrossRef CAS PubMed.
- D. Holzmann, D. Holzinger, G. Hesser, T. Schmidt and G. Knör, J. Mater. Chem., 2009, 19, 8102–8106 RSC.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS PubMed.
- F. Caruso, M. Spasova, V. Salgueiriño-Macieira and L. M. Liz-Marzán, Adv. Mater., 2001, 13, 1090–1094 CrossRef CAS.
- Z. Wang, S. Zong, J. Yiang, J. Li and Y. Cui, Biosens. Bioelectron., 2011, 26(6), 2883–2889 CrossRef CAS PubMed.
- M. Bradley and B. S. Garcia-Risueño, J. Colloid Interface Sci., 2011, 355(2), 321–327 CrossRef CAS PubMed.
- D.-Q. Yang, M. Meunier and E. Sacher, J. Appl. Phys., 2004, 95, 5023–5026 CrossRef CAS.
- K. van Werde, D. Mondelaers, G. Vanhoyland, D. Nelis, M. K. van Bael, J. Mullens and L. C. van Poucke, J. Mater. Sci., 2002, 37, 81–88 CrossRef CAS.
- S. Kumar, K. S. Gandhi and R. Kumar, Ind. Eng. Chem. Res., 2004, 46(10), 3128–3136 CrossRef.
- S. Ivanova, C. Petit and V. Pitchon, Appl. Catal., A, 2004, 267, 191–201 CrossRef CAS.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231(1), 105–114 CrossRef CAS.
- K. Zhao, B. Qiao, J. Wang, Y. Zhang and T. Zhang, Chem. Commun., 2011, 47, 1779–1781 RSC.
- J.-D. Wang, J.-K. Liu, Y. Lu, D.-J. Hong and X.-H. Yang, Mater. Res. Bull., 2014, 55, 190–195 CrossRef CAS.
- A. J. Mieszawska, W. J. M. Mulder, Z. A. Fayad and D. P. Cormode, Mol. Pharmaceutics, 2013, 10(3), 831–847 CrossRef CAS PubMed.
- J. Park, J. Park, E. J. Ju, S. S. Park, J. Choi, J. H. Lee, K. J. Lee, S. H. Shin, E. J. Ko, I. Park, C. Kim, J. J. H. Wang, J. S. Lee, S. Y. Song, S.-Y. Jeong and E. K. Choi, J. Controlled Release, 2015, 207, 77–85 CrossRef CAS PubMed.
- T. Scharnweber, C. Santos, R.-P. Franke, M. M. Almeida and M. E. V. Costa, Molecules, 2008, 13, 2729–2739 CrossRef CAS PubMed.
- Y. Liu, X. Cao, M. Luo, Z. Le and W. Xu, J. Colloid Interface Sci., 2009, 329(2), 244–252 CrossRef CAS PubMed.
- J. P. Bilezikian, L. G. Raisz and T. J. Martin, Principles of Bone Biology, Academic Press, vol. 1, 2008 Search PubMed.
- I. Titushkin, S. Sun, J. Shin and M. Cho, J. Biomed. Biotechnol., 2010, 2010, 1–14 CrossRef PubMed.
- R. Gonzalez-McQuire, D. W. Green, K. A. Partridge, R. O. C. Oreffo, S. Mann and S. A. Davis, Adv. Mater., 2007, 19, 2236–2240 CrossRef CAS.
- Y. Cai and R. Tang, J. Mater. Chem., 2008, 18, 3775–3787 RSC.
- Z. Shi, X. Huang, Y. Cai, R. Tang and D. Yang, Acta Biomater., 2009, 5(1), 338–345 CrossRef CAS PubMed.
- J. L. Xu, K. A. Khor, J. J. Sui, J. H. Zhang and W. N. Chen, Biomaterials, 2009, 30(29), 5385–5391 CrossRef CAS PubMed.
- Y. Liu, G. Wang, Y. Cai, H. Ji, G. Zhou, X. Zhao, R. Tang and M. Zhang, J. Biomed. Mater. Res., Part A, 2009, 90, 1083–1091 CrossRef PubMed.
- T. Mustafa, F. Watanable, W. Monroe, M. Mahmood, Y. Xu, L. Saeed, A. Karmakar, D. Casciano, S. Ali and A. Biris, J. Nanomed. Nanotechnol., 2011, 2(6), 118–124 Search PubMed.
- D. D. Liu, J. C. Zhang, C. Q. Yi and M. S. Yang, Chin. Sci. Bull., 2010, 55, 1013–1019 CrossRef.
- C. Yi, D. Liu, C.-C. Fong, J. Zhang and M. Yang, ACS Nano, 2010, 4, 6439–6448 CrossRef CAS PubMed.
- J.-H. Fan, W.-T. Li, W.-I. Hung, C.-P. Chen and J.-M. Yeh, Biomed. Eng., 2011, 23, 141–152 CAS.
- W. Cui, J. Li, Y. Zhang, H. Rong, W. Lu and L. Jiang, Nanomedicine: Nanotechnology, Biology, and Medicine, 2012, 8(1), 46–53 CrossRef CAS PubMed.
- J. A. Yang, S. E. Lohse and C. J. Murphy, Small, 2014, 10, 1642–1651 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |