
A prelithiated lithium vanadate anode and the mass balancing of its hybrid capacitor
Abstract
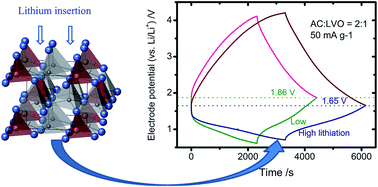
The electrode of sol–gel derived Li3VO4 (LVO), which exhibits a capacity of 2 mol lithium between 0.01 and 2.0 V (vs. Li/Li+), is studied as an anode of the capacitor in conjunction with a cathode of activated carbon (AC). Before assembling the hybrid capacitor of AC/LVO, Li3VO4 has been prelithiated. The end potential of prelithiation affects the lithium loading and the resultant cell capacity. The cell, with LVO prelithiation ending at 0.5 V (vs. Li/Li+), displays 34% more capacity than the other with LVO ending at 2.0 V (vs. Li/Li+). The higher capacity is caused by reducing the electrode potential of the hybrid capacitor at 0% state-of-charge, which also depends on the AC : LVO mass ratio and the specific current. We develop a scheme to estimate the optimal mass ratio for the capacitor. The optimal ratio is verified later by galvanostatic charge–discharge experiments, involving four capacitors with AC : LVO mass ratios of 0.5 : 1, 1 : 1, 2 : 1, 3 : 1. The optimal 2 : 1 cell demonstrates a capacity of specific energy 49.1 W h kg−1 at 0.05 A g−1 and 3.5 V, and 81% retention in the voltage hold test persisting for 100 h.
 Please wait while we load your content...
Please wait while we load your content...

