
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of cyclobutane thymine dimers by tiaprofenic acid and its photoproducts: approach to the photosensitizer triplet state energy limit value
Sandra
Michaud
ab,
Guillaume
Bordeau
ab,
Valérie
Sartor
ab,
Jose Luis
Bourdelande
c,
Jordi
Hernando
c,
Gonzalo
Guirado
c and
Nadia
Chouini-Lalanne
*ab
aLaboratoire des Interactions Moléculaires et Réactivité Chimique et Photochimique (IMRCP), UMR 5623, Université Paul Sabatier, Bât 2R1, 118, route de Narbonne, 31062 Toulouse Cedex, France. E-mail: lalanne@chimie.ups-tlse.fr
bCNRS, Laboratoire IMRCP, UMR 5623, 31062 Toulouse Cedex 09, France
cDepartement de Quimica, Universitat Autonoma de Barcelona, 08193-Bellaterra, Barcelona, Spain
First published on 4th August 2015
Abstract
Cyclobutane thymine dimers, the major photoproducts produced in UV-irradiated DNA, are the main causative agents for mutagenesis and skin cancer. This lesion can also be initiated under UVA radiation, involving triplet–triplet energy transfer mechanism from a photosensitizer to the thymine nucleobase. According to previous reports, only photosensitizers with a triplet state energy >270 kJ mol−1 should be able to induce cyclobutane thymine dimers photosensitization. However, tiaprofenic acid, a non-steroidal anti-inflammatory drug widely prescribed in the treatment of inflammation and pain, has shown cyclobutane thymine dimers photosensitization, although its triplet energy state value and those of its photoproducts are lower than the one previously reported for thymine in DNA. In this context, the in vitro photosensitizing properties of tiaprofenic acid and its photoproducts were studied by agarose gel electrophoresis and phosphorescence experiments and demonstrated clearly the formation of cyclobutane thymine dimers by tiaprofenic acid and its photoproducts. This study allows us to approach the lower limit threshold of the triplet state energy of a photosensitizer for cyclobutane thymine dimers formation and thereby to improve the prediction of the photogenotoxic potential of current and future drugs.
Introduction
UVB radiation is one of the most important environmental factors causing alterations of chemical integrity of DNA, especially on adjacent pyrimidine bases. It leads to the formation of dimeric photoproducts, mainly cyclobutane thymine dimers (CPD) at the origin of biological effects such as mutagenesis and skin cancer.1–9 This major lesion, which is formed by a [2 + 2] photocycloaddition reaction of adjacent thymines, can also be initiated upon DNA photosensitization involving a triplet–triplet energy transfer mechanism from a photosensitizer to thymine nucleobase.5–8,10–16 To be efficient, the photosensitizer must combine a good intersystem crossing quantum yield (ΦISC) with a triplet state energy (ET) higher than the one of the thymine, which is the mononucleotide having the lowest triplet state. This critical energy value was determined for thymine in solution at 310 kJ mol−1.10,11 Some fluoroquinolone DNA photosensitizers have been used to probe the ET of thymine in DNA and the triplet state energy of thymine in pBR322 plasmidic DNA has been established at 270 kJ mol−1.17,18 This value has been estimated by laser flash photolysis experiments of phosphate buffer solutions of fluoroquinolones, norfloxacin (NFX, ET = 269 kJ mol−1) and its N(4′) acetyl derivative (ANFX, ET = 265 kJ mol−1) using triplet–triplet energy transfer experiments.17 The study of the photosensitizing properties of these two compounds to pBR322 plasmidic DNA showed that NFX photosensitized cyclobutane thymine dimers while ANFX not.If this approach is interesting, it is not always sufficient in particular for photochemically unstable photosensitizers. Indeed, the formation of thymine dimers can result of energy transfer either from the parent drug or from its photoproducts.14,19 So, the role of drug photoproducts in thymine dimer photosensitization has to be considered and their triplet state energies determined to estimate the lower limit threshold of a photosensitizer triplet state energy to excite the triplet state of thymine leading to the CPD formation.
In this context, we chose to investigate the DNA photosensitizing properties of tiaprofenic acid, a non-steroidal anti-inflammatory drug and its three major photoproducts: ethyl (EthylTP), hydroxyethyl (HydroxyTP) and acetyl (AcetylTP) derivatives (Scheme 1). This drug, which is among the most widely prescribed in the treatment of inflammation and pain, has been repeatedly associated with photosensitivity secondary effects.20,21 Its photochemical and photobiological properties towards biological targets such as cell membranes or DNA have been investigated22,23 and cyclobutane thymine dimer photosensitization was demonstrated upon TP photolysis.12
 | ||
| Scheme 1 Chemical structures of tiaprofenic acid and its photoproducts. | ||
According to previous reports, only drugs with ET > 270 kJ mol−1 could induce cyclobutane thymine dimer photosensitization.17,18,24 For tiaprofenic acid (TP), the photoreactive excited state is the lowest triplet state T1 (ref. 25–28) with an energy value determined to be 242 kJ mol−1 by phosphorescence experiment.29,30 Hence, it was reasonable to expect that during TP photolysis, the formation of such damage must be due to photoproducts rather than the parent drug itself. Accordingly it means that, at least for one of TP photoproducts (Scheme 1), ET should be > 270 kJ mol−1. However, in a recent study, the photophysical properties of TP photoproducts were investigated.30 The results, obtained by phosphorescence emission at 77 K in an ethanol glass, show for all photoproducts ET < 242 kJ mol−1 (Table 1). Therefore, since the formation of cyclobutane thymine dimers was demonstrated during TP photolysis, it is important from a drug safety point of view, to confirm and to verify the photosensitivity of CPD by TP and its photoproducts in order to approach the ET allowing CPD photosensitization in DNA, an important data for predicting photogenotoxic potential of current and future drugs.
| Photosensitizers | TP | AcetylTP | HydroxyTP | EthylTP |
|---|---|---|---|---|
| a Determined by phosphorescence experiments at low temperature (77 K) in an ethanol glass (λexc = 314 nm).30 b Determined by phosphorescence experiments at 25 °C in buffered aqueous solutions (λexc = 355 nm). c Estimated values for TTET experiments from phosphorescence experiments. | ||||
| E T a (kJ mol−1) | 242 | 221 | 242 | 238 |
| E T b (kJ mol−1) | 221 | 217 | 221 | 225 |
| E T c (kJ mol−1) | 254 | 233 | 254 | 250 |
Results and discussion
Phosphorescence experiments
The triplet state energies of TP photoproducts recently reported have been determined from the emission phosphorescence experiments in ethanol at 77 K.30 However, in order to assess the triplet state energy value of photosensitizers in biological conditions, emission phosphorescence experiments were carried out in buffered aqueous solution at room temperature (Table 1). For this purpose, AcetylTP, HydroxyTP and EthylTP were synthesized by TP photolysis carried out by sunlight irradiation of a phosphate buffered solution of TP, purified by column chromatography30 and their triplet state energies were evaluated by phosphorescence emission experiments in buffered aqueous solutions at controlled temperature (25 °C) (Fig. 1). It appears clearly that lower values of ET were obtained from the measurements in buffer aqueous solutions at controlled temperature (∼5–20 kJ mol−1). This can be ascribed to an additional stabilization of the triplet emissive state arising from solvation leading to underestimate the actual values of the energies of triplet states. Moreover, an important feature of the phosphorescence spectra in Fig. 1 is that they are rather broad, which indicates that a variety of triplet energies might be available for these photosensitizers. Indeed, considering the blue-shifted tail of these spectra, the triplets as high energy as ∼270 kJ mol−1 and ∼250 kJ mol−1 can be estimated for EthylTP and the rest of the products respectively, suggesting that the evaluation of triplet energy of these compounds directly by the phosphorescence spectral maximum, might provide with a lower-limit value. For instance, in the case of norfloxacin (NFX), one of the fluoroquinolone DNA photosensitizers used to determine ET of thymine in DNA, ET was found to be 257 kJ mol−1 from its phosphorescence spectral maximum at 77 K in contrast to the 269 kJ mol−1 upper limit value established from triplet–triplet energy transfer experiments (TTET).18,31 Using for TP and its photoproducts, the ET values derived from phosphorescence measurements at 77 K and assuming a similar difference, ET in the range of ∼233–255 kJ mol−1 could be estimated for TP family, which is in reasonably good agreement with the highest energy region of their phosphorescence spectra (Table 1). As such, this will be taken into account in the forthcoming discussion.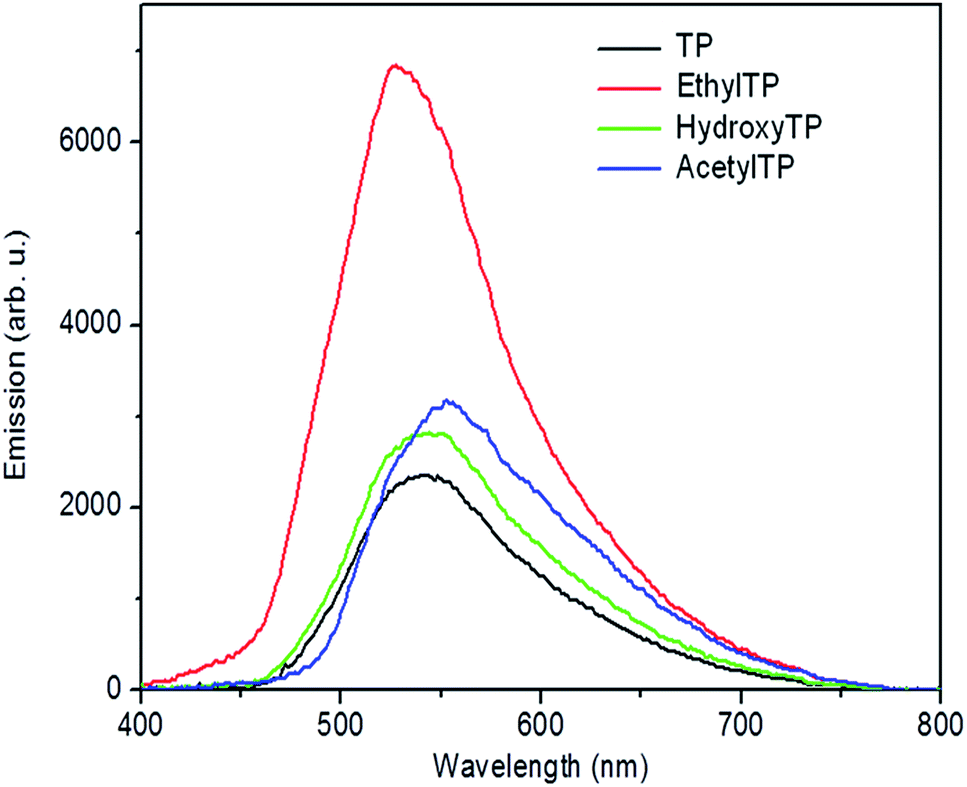 | ||
| Fig. 1 Phosphorescence emission spectra of TP and its photoproducts in buffered aqueous solutions at 25 °C (λexc = 355 nm). | ||
Photosensitization of cyclobutane thymine dimers by TP and its photoproducts
The ability to induce cyclobutane thymine dimers was investigated on tiaprofenic acid and all its photoproducts and compared to riboflavin, a drug with ET = 209 kJ mol−1 (from phosphorescence experiments at 77 K), well known to mediate photooxidation DNA damage both by electron and energy transfer.32–34 CPD photosensitization by riboflavin has never been reported. The experiments were carried out at 20 °C and on plasmidic DNA pBR322, a very useful tool to detect DNA damages, irradiated at λ > 320 nm in presence of drugs in phosphate buffered solution at room temperature. Thymine dimer formation was detected using phage T4 endonuclease V, a DNA repair enzyme that selectively excises cyclobutane pyrimidine dimers by single-strand DNA scission. Revelation of these single-strand breaks was possible using conversion of pBR322 plasmidic DNA from the supercoiled form (or Form I) to the circular form (or Form II) separated by agarose gel electrophoresis and quantified by photodensitometry. The results are shown in Fig. 2 and 3.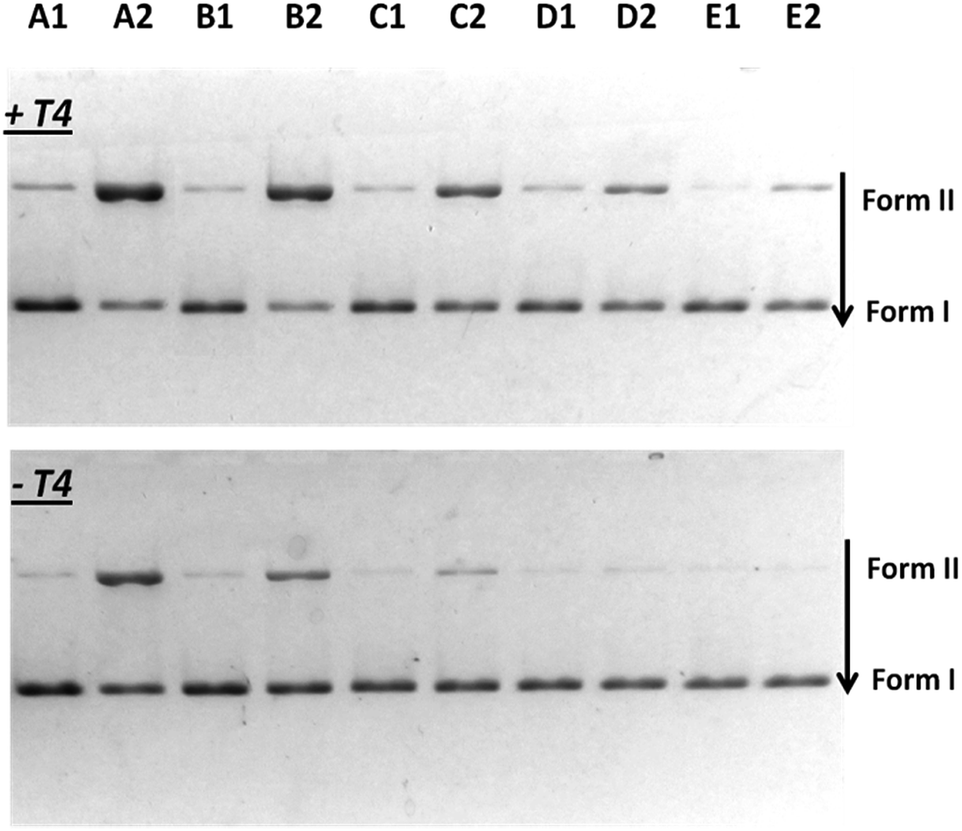 | ||
| Fig. 2 Agarose gel showing CPD photosensitization by TP and its photoproducts. pBR322 plasmidic DNA (20 μM in base pairs) submitted (+T4) or not (−T4) to T4 endonuclease treatment to reveal thymine dimers formation after UVA irradiation of samples at λ > 320 nm for 2 min. Form I corresponds to the supercoiled structure and Form II to the circular structure. Lane E1: non irradiated alone; Lane E2 irradiated alone; Lane A1: non irradiated in presence of 40 μM of TP; Lane A2: irradiated in presence of 40 μM of TP; Lane B1: non irradiated in presence of 40 μM of HydroxyTP; Lane B2: irradiated in presence of 40 μM of HydroxyTP; Lane C1: non irradiated in presence of 40 μM of AcetylTP; Lane C2: irradiated in presence of 40 μM of AcetylTP; Lane D1: non irradiated in presence of 40 μM of EthylTP; Lane D2: irradiated in presence of 40 μM of EthylTP. | ||
 | ||
| Fig. 3 Percentage of photo-induced DNA damage photosensitized by TP and TP photoproducts: cyclobutane thymine dimers (light grey) and single strand breaks (dark grey) at 20 °C. | ||
As seen in Fig. 2, when plasmidic DNA is irradiated in presence of TP and of each TP photoproducts and without enzymatic treatment (−T4), single-strand breaks corresponding to a Type I radical mechanism were observed in particular for TP, HydroxyTP and AcetylTP (Lane A2, B2, C2) in agreement with previous work.30 Irradiation of pBR322 plasmidic DNA in presence of TP and its photoproducts followed by endonuclease treatment (+T4) led for each of these compounds, to the formation of cyclobutane thymine dimers (Lane A2, B2, C2, D2). These results demonstrate unambiguously for TP and its all photoproducts, the involvement of efficient triplet–triplet energy transfer from drug to the thymine base at the origin of the photosensitization of cyclobutane thymine dimers.
A quantitative analysis of the cyclobutane thymine dimers photosensitization by these drugs has been done by photodensitometry (Fig. 3).
Cyclobutane thymine dimers photosensitization occurs by triplet–triplet energy transfer. Then, triplet state energy and intersystem crossing quantum yield of the photosensitizer are critical parameters in this process, which modulate their efficiency and ability on thymine dimer formation. The results obtained by the quantitative analysis suggest that HydroxyTP is the strongest photosensitizer (Fig. 3). This is probably associated to a higher triplet state energy than the other photoproducts, combined with an efficient intersystem crossing quantum yield. In the case of EthylTP, its lower photosensitization efficiency could be correlated to a much less effective intersystem crossing quantum yield. For AcetylTP, although it has the lowest energy, its effective cyclobutane thymine dimers photosensitization is probably due to an efficient intersystem crossing quantum yield due to the attachment of the acetyl substituent to the benzoylthiophene moiety, a heterocyclic benzophenone, which should greatly enhance intersystem crossing quantum yield.
In order to eliminate the possibility of protein promoting base catalyzed cleavage at oxidative lesions, a control with heat-denatured enzyme has been also run with EthylTP. It led to the same pattern than that obtained with EthylTP without enzymatic treatment, namely a production of single-strand breaks (Fig. 4). Moreover, when pBR322 plasmidic DNA is irradiated in presence of riboflavin followed by endonuclease treatment, no formation of cyclobutane thymine dimers photosensitized by riboflavin is observed (Fig. 4).
 | ||
| Fig. 4 Percentage of photo-induced DNA damage photosensitized by EthylTP with normal and denaturated T4 enzyme and by riboflavin: cyclobutane thymine dimers (light grey) and single strand breaks (dark grey). | ||
All these results highlight for TP and its photoproducts a triplet energy available for CPD formation in the range of ∼233–255 kJ mol−1, indicating a significant difference with the triplet state energy for CPD photosensitization given in the literature.17 The energy gap between estimated triplet energies of TP and its photoproducts, and the triplet state energy value for CPD formation given in the literature (ΔET ranges from 15 kJ mol−1 to 35 kJ mol−1), does not permit to speculate the possibility of thermally activated triplet–triplet energy transfer from populated vibrationally excited levels of photosensitizers. Actually, to investigate possible thermal effects on CPD formation, additional photo-induced DNA damage studies were carried out at a lower temperature (5 °C) for the TP photoproducts showing the largest (HydroxyTP) and smallest (EthylTP) effect at 20 °C. Noticeable, both compounds were also found to promote CPD formation at 5 °C, with efficiencies comparable to those observed at 20 °C. Thus, photosensitization with HydroxyTP yielded 85% of the cyclobutane thymine dimers determined at room temperature, whereas 55% of them were formed with EthylTP. It must be noted that high-lying triplet states of TP and its derivatives should not be significantly populated at the low temperature considered according to the T2 energy reported for TP,29 which further suggests that DNA photosensitization should proceed via energy transfer from T1. Indeed, this is in agreement with recent reports about the photoreactivity of TP.25–28
In addition, these low temperature experiments reinforce our finding the photosensitization of CPD formation for TP and its photoproducts can take place at energies lower than the 270 kJ mol−1 value previously reported.17 In fact, recent CASPT2 predictions showed that the threshold energy for activating the photosensitization mechanism to form cyclobutane pyrimidine dimers is not the energy of the equilibrium structure of the lowest-lying triplet excited state of the nucleobase (2.87 eV, 277 kJ mol−1) but rather the relevant energy of the singlet–triplet crossing intermediary structure of biradical character (2.36 eV, 227.7 kJ mol−1) that connects the excited dimer with the cylobutane pyrimidine product.35,36 Our results indeed demonstrate that lower energies than 270 kJ mol−1 promote CPD formation since they prove unequivocally that TP and its photoproducts, with an ET in the range of ∼233–255 kJ mol−1, excite the triplet state of thymine leading to the photosensitization of CPD (Fig. 5). Such finding could have serious consequences for drug safety and for the determination of triplet energy values of photosensitizers in the condensed phase.
 | ||
| Fig. 5 Lower limit threshold of a photosensitizer triplet state energy (in kJ mol−1 and eV) for CPD formation (from estimated values for TTET). | ||
Conclusions
In conclusion, we demonstrate in this work that assessment of drug effects on DNA photodamage evolving through thymine dimerization requires the study of the photosensitization efficiency of both the parent therapeutic agent and its photoproducts. In particular, we observed in our study that such a process occurs under the influence of not only tiaprofenic acid, but also of its photodegradation products displaying lower triplet state energies such as AcetylTP.These results allow us to approach in condensed phase, the lower limit threshold of a photosensitizer triplet state energy to excite the triplet state of thymine leading to the photosensitization of CPD. From the drug safety point of view, the knowledge of this key parameter in cyclobutane thymine dimer photosensitization is of fundamental interest for predicting photogenotoxic potential of current and future drugs.
Experimental part
Materials
Tiaprofenic acid (TP) (2-(5-[2-benzoyl]thienyl)propionic acid) was extracted from Flanid (Pierre Fabre medicament production, Boulogne, France). TP photoproducts (2-benzoyl-5-ethylthiophene (EthylTP), 2-benzoyl-5-(1-hydroxyethyl)thiophene (HydroxyTP) and 2-benzoyl-5-acetylthiophene (AcetylTP)) were obtained by TP photolysis carried out by sunlight irradiation of a phosphate buffered solution of TP, until the drug had totally consumed and purification by column chromatography, according to a reported procedure.26 Ethidium bromide (EB) as a 10 mg mL−1 water solution was purchased from Pharmacia Biotech Plusone. Phage T4 endonuclease V was purchased from Epicentre. Supercoiled plasmid DNA (Form I) pBR322 (4361 base pairs) was purchased from Thermo Scientific. The amount of contaminant relaxed circular DNA (Form II) was controlled by agarose gel electrophoresis followed by photodensitometry and was less than 10%. No linear DNA (Form III) was detected in the starting material.Phosphorescence experiments
Solutions were prepared in PBS buffer with a ∼5% content in MeOH to ensure complete dissolution of the compounds and an OD ∼ 0.1 at the excitation wavelength. Phosphorescence measurements were registered in a custom-made spectrofluorometer where the 3rd harmonic (355 nm) of a pulsed Nd:YAG laser (Brilliant, Quantel, pulse width ∼ 10 ns, pulse energy ∼ 3 mJ) was used to excite the Ar-saturated drug solutions in a temperature-controlled, 1 cm quartz cuvette. Resulting emission was spectrally resolved in a spectrograph and finally detected in an intensified CCD camera (iStar, Andor). Collected signal was integrated for t = 1 μs to 10 ms. The triplet energies were then determined from the maxima of the measured phosphorescence spectra.Photosensitization experiments and detection of cyclobutane thymine dimers
All the solutions were prepared in 50 mM Tris/HCl buffer (5 mM EDTA, 5 mM MgCl2, pH = 7.5). Samples containing pBR322 plasmidic DNA (20 μM base pair) alone or in presence of photosensitizer (40 μM, TP or HydroxyTP or AcetylTP or EthylTP) were irradiated 2 min at 20 °C using a 500 W Hg/xenon lamp (Oriel) equipped with a long pass filter λ > 320 nm. After irradiation and photosensitizer removal using Amicon Ultra centrifugal filters (3 K membrane, Millipore), samples were splitted into two batches. For each sample, one of them was incubated for 30 min at 37 °C with an excess of T4 endonuclease V.Agarose gel electrophoresis
Prior electrophoresis, 5 μL of loading buffer (250 mM Hepes, pH 7.45, 75% glycerol, and 0.005% bromophenol blue) was added to each sample. An aliquot of 30 μL of each solution was directly loaded into 0.8% agarose gel, containing ethidium bromine and electrophoresis was conducted under a constant electric field of 20 V overnight (circa 16h). Photodensitometry of the resulting gels was carried out with ImageJ software (National Institute of Health). All damage experiments were carried out at least three times.Acknowledgements
This work was supported by grant CTQ2012-30853 from “Ministerio de Economía y Competitividad” (MINECO).Notes and references
- J. Cadet, E. Sage and T. Douki, Mutat. Res., 2005, 571, 3–17 CrossRef CAS PubMed.
- Y. Matsumura and H. N. Ananthaswamy, J. Appl. Pharmacol., 2004, 195, 298–308 CrossRef CAS PubMed.
- A. Gentil, F. le Page and A. Sarasin, Biol. Chem., 1997, 378, 1287–1292 CAS.
- J. S. Taylor, Pure Appl. Chem., 1995, 67, 183–190 CrossRef CAS.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS PubMed.
- M. Charlier, C. Helene and W. L. Carrier, Photochem. Photobiol., 1972, 15, 527–536 CrossRef CAS.
- A. A. Lamola, Pure Appl. Chem., 1970, 24, 599–610 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS PubMed.
- J. S. Taylor, Acc. Chem. Res., 1994, 27, 76–82 CrossRef CAS.
- P. D. Wood and R. W. Redmond, J. Am. Chem. Soc., 1996, 118, 4256–4263 CrossRef CAS.
- I. G. Gut, P. D. Wood and R. W. Redmond, J. Am. Chem. Soc., 1996, 118, 2366–2373 CrossRef CAS.
- N. Chouini-Lalanne, M. Defais and N. Paillous, Biochem. Pharmacol., 1998, 55, 441–446 CrossRef CAS.
- V. Lhiaubet-Vallet, J. Trzcionka, S. Encinas, M. A. Miranda and N. Chouini-Lalanne, Photochem. Photobiol., 2003, 77, 487–491 CrossRef CAS.
- V. Lhiaubet-Vallet, J. Trzcionka, S. Encinas, M. A. Miranda and N. Chouini-Lalanne, J. Phys. Chem. B, 2004, 108, 14148–14153 CrossRef CAS.
- S. Sauvaigo, T. Douki, F. Odin, S. Caillat, J. L. Ravanat and J. Cadet, Photochem. Photobiol., 2001, 73, 230–237 CrossRef CAS.
- L. Marrot, J. P. Belaidi, C. Jones, P. Perez, L. Riou, A. Sarasin and J. R. Meunier, J. Invest. Dermatol., 2003, 121, 596–606 CrossRef CAS PubMed.
- F. Bosca, V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell and M. A. Miranda, J. Am. Chem. Soc., 2006, 128, 6318–6319 CrossRef CAS PubMed.
- V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell, F. Bosca and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 7409–7414 CrossRef CAS PubMed.
- J. Trzcionka, V. Lhiaubet-Vallet and N. Chouini-Lalanne, Photochem. Photobiol. Sci., 2004, 3, 226–230 CAS.
- B. Ljunggren, Photodermatology, 1985, 2, 3–9 CAS.
- I. E. Kochevar, Arch. Dermatol., 1989, 125, 824–826 CrossRef.
- G. Condorelli, G. de Guidi, S. Giuffrida and L. L. Costanzo, Coord. Chem. Rev., 1993, 125, 115–128 CrossRef CAS.
- F. Bosca, M. Luisa Marin and M. A. Miranda, Photochem. Photobiol., 2001, 74, 637–655 CrossRef CAS.
- C. Cuquerella, V. Lhiaubet-Vallet, J. Cadet and M. A. Miranda, Acc. Chem. Res., 2012, 45, 1558–1570 CrossRef PubMed.
- F. Bosca and M. A. Miranda, J. Photochem. Photobiol., B, 1998, 43, 1–26 CrossRef CAS.
- M. A. Miranda, J. V. Castell, D. Hernandez, M. J. Gomez-Lechon, F. Bosca, I. M. Morera and Z. Sarabia, Chem. Res. Toxicol., 1998, 11, 172–177 CrossRef CAS PubMed.
- A. Klefah, K. Musa and L. A. Eriksson, J. Phys. Chem. B, 2009, 113, 11306–11313 CrossRef PubMed.
- T. Su, J. Ma, M.-D. Li, X. Guan, L. Yu and D. L. Phillips, J. Phys. Chem. B, 2013, 117, 811–824 CrossRef CAS PubMed.
- S. Encinas, M. A. Miranda, M. Giancarlo and S. Monti, Photochem. Photobiol., 1998, 67, 420–425 CrossRef CAS.
- S. Michaud, V. Hajj, L. Latapie, A. Noirot, V. Sartor, P. L. Fabre and N. Chouini-Lalanne, J. Photochem. Photobiol., B, 2012, 110, 34–42 CrossRef CAS PubMed.
- L. Martínez, P. Bilski and C. F. Chignell, Photochem. Photobiol., 1996, 64, 911–917 CrossRef.
- R. W. Chambers and D. R. Kearns, Photochem. Photobiol., 1969, 10, 215–219 CrossRef CAS.
- W. Lin, C. Lu, F. Du, Z. Shao, Z. Han, T. Tu, S. Yao and N. Lin, Photochem. Photobiol. Sci., 2006, 5, 422–425 CAS.
- G. T. Wondrack, K. M. Jacobson and E. L. Jacobson, Photochem. Photobiol. Sci., 2006, 5, 215–237 Search PubMed.
- T. Climent, I. Gonzalez-Ramírez, R. Gonzalez-Luque, M. Merchan and L. Serrano-Andres, J. Phys. Chem. Lett., 2010, 1, 2072–2076 CrossRef CAS.
- D. Roca-Sanjuán, G. Olaso-González, I. González-Ramírez, L. Serrano-Andrés and M. Merchán, J. Am. Chem. Soc., 2008, 130, 10768–10779 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |