
Electrochemical detection of mercury using biosynthesized hydroxyapatite nanoparticles modified glassy carbon electrodes without preconcentration†
Abstract
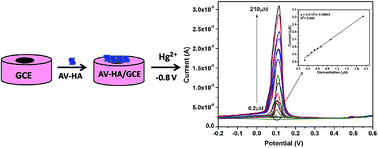
An electrochemical method for the determination of trace levels of mercury(II) ions using hydroxyapatite (HA) nanoparticles modified glassy carbon electrode (GCE) by square wave voltammetry is described for the first time. HA nanoparticles biosynthesized using Aloe vera plant (Av) extract exhibited improved electrocatalytic activity towards Hg2+ ions when compared to pristine HA. The Av-HA modified GCE showed an excellent selectivity and improved sensitivity towards the detection of Hg2+ without requiring preconcentration of mercury during voltammetric detection. Operational parameters such as pH, supporting electrolyte and scanning potential range were optimized. Under the optimum experimental conditions, the anodic peak current is proportional to the concentrations of mercury over a wide range of 2.0 × 10−7 to 2.1 × 10−4 M with the lowest detectable concentration of 141 nM. The Av-HA modified GCE showed good selectivity towards mercury in the presence of potential interferents such as copper, lead, cadmium, silver and zinc. The fabricated sensor displayed good reproducibility and is suitable for the determination of mercury in tap water and industry waste water.
 Please wait while we load your content...
Please wait while we load your content...

