
Ethylenediamine-functionalized magnetic Fe3O4@SiO2 nanoparticles: cooperative trifunctional catalysis for selective synthesis of nitroalkenes†
Fengjun Xueab,
Yahao Dongab,
Peibo Huab,
Yanan Dengab and
Yuping Wei*ab
aDepartment of Chemistry, School of Science, Tianjin University, Tianjin 300072, P. R. China. E-mail: ypwei@tju.edu.cn; Fax: +86 22 27403475; Tel: +86 22 27403475
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R. China
First published on 25th August 2015
Abstract
A magnetically separable trifunctional nanocatalyst Fe3O4@SiO2–NH2 was synthesized and characterized by TEM, FT-IR, XRD, TGA, and EA. The designed nanocatalyst was found to be highly active for selective synthesis of nitroalkenes with nitromethane and aromatic aldehyde through cooperative trifunctional catalysis of primary amine, secondary amine and Si–OH groups on the surface of the catalyst. Under the optimized conditions, various representative substrates were extended to obtain the corresponding products in moderate or excellent yields. After the reaction, the trifunctional nanocatalyst was easily recovered and recycled by applying an external magnet. In addition, a possible cooperative trifunctional catalysis mechanism was also proposed.
1 Introduction
Since the concept of a monomolecular bifunctional catalyst for cooperative catalysis was first proposed in 2003 by Takemoto group,1 both homogeneous and heterogeneous catalysts with molecular design and their application in organic transformations have become the focus of attention. The pursuit of green chemical processes impels scientists and researchers to exploit more recoverable catalysts with multifunctional surfaces. The immobilization of homogeneous catalysts on solid supports has been considered as an efficient approach to realize catalyst recycling.2 It is worth mentioning that the dispersion of most heterogeneous catalysts in reaction medium are poor.3,4 Therefore, the catalytic activity of the heterogeneous catalyst will be inevitably affected compared to the corresponding homogeneous catalyst. Due to the large specific surface area and versatile surface, nanoparticles have attracted much attention for immobilization of homogeneous catalysts.5 However, nanoparticles are difficult to separate by conventional filtration techniques.6 Magnetic nanoparticles are a promising option.6 As an ideal support, magnetic Fe3O4 is non-toxic, easy to prepare, active for modification and functionalization.5 From the point view of sustainable development, magnetically separable nanocatalysts with multifunctional surface are attracting researches' great attentions.Nitroalkenes are versatile building blocks7 because they have been widely used as substrates such as Michael addition,1,8 Friedel–Crafts alkylation,9,10 1,3-dipolar cycloaddition11,12 and domino reaction.13 They have also been used as key precursors of different targets for scavengers of macrophage-generated oxidants,14 antidepressant drugs15 and rat even human monoamine oxidase inhibitors.16 The nitro group can be converted into various functional groups.17,18 Several reports about nitroalkenes synthesis have been described.17–26 The immobilization of amines on silica has received much attention due to the large specific surface area and good dispersion.19–23 In these reports, few nanocatalysts have bifunctional even trifunctional active sites on their surfaces.21,22 Additionally, conventional filtration and centrifuge for isolation of these silica-supported nanocatalysts from the reaction mixture are not so efficient.6 Magnetic Fe3O4-supported catalysts with multifunctional surface not only can achieve simple and efficient separation but also can collaboratively accelerate a single reaction or enable one-pot reaction sequences.
With this background in mind, in this paper a magnetically separable trifunctional nanocatalyst (Scheme 1, Fe3O4@SiO2–NH2) was prepared for cooperative catalytic synthesis of nitroalkenes under mild conditions without additional solvents. The reaction between nitromethane and aromatic aldehyde might experience a cooperative trifunctional catalytic process which was promoted by primary amine, secondary amine and Si–OH groups on the surface of Fe3O4@SiO2–NH2. Under the optimized conditions, various representative substrates were extended. After reaction the separation and recovery of magnetic Fe3O4 supported catalysts could be achieved using an external magnet. Combined with the experimental data and literatures, possible reaction mechanism for selective synthesis of nitroalkenes through a trifunctional cooperative catalysis was proposed.17–19,21,22,25–27
 | ||
| Scheme 1 The synthesis of Fe3O4@SiO2–NH2. | ||
2 Experimental section
2.1 Materials
Ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), ammonia (25 wt%), trisodium citrate, acetonitrile (CH3CN), ethanediamine (NH2CH2CH2NH2) were purchased from Tianjin Guangfu Fine Chemical Research Institute (Tianjin, P. R. China). Toluene (C6H5CH3) was obtained from Tianjin Jiangtian Chemical Technology Co., Ltd. Absolute alcohol, nitromethane were supplied by Tianjin Real & Lead Chemical Co., Ltd. Tetraethyl orthosilicate (TEOS) was obtained from Tianjin Kemiou Chemical Reagent Co., Ltd. Chloropropyl triethoxysilane was purchased from Heowns Biochem Technologies LLC. Dichloromethane (CH2Cl2) and toluene (C6H5CH3) were dried with CaH2, distilled under reduced pressure and stored over molecular sieves. Other reagents, unless otherwise stated, were used as received for the reaction without further purification. Deionized water was used for all experiments.2.2 Analytical methods
NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer in CDCl3 using TMS as an internal standard. The morphology of the samples was observed on a Tecnai G2 F20 transmission electron microscopy (TEM). The content of nitrogen was determined by a Vario EL cube elemental analyzer. Thermogravimetric analysis (TGA) was measured with a STA 409 PC thermal analyzer (NETZSCH, Germany). Fourier transform infrared spectroscopy (FTIR) analysis was performed on a BIO-RAD FTS3000 IR Spectrum Scanner. The X-ray diffraction (XRD) spectra of the samples were measured using an X-ray diffractometer (BDX3300) with reference target: Cu Kα radiation (λ = 1.54 Å), voltage: 30 kV and current: 30 mA. The samples were measured from 10 to 90° (2θ) with steps of 4° min−1. Preparative TLC (20 cm × 20 cm) was performed on Silica Gel 60 F254.2.3 Preparation of Fe3O4 magnetic nanoparticles (Fe3O4 MNPs)
The Fe3O4 MNPs were prepared following the reported chemical co-precipitation methods with slight modification.28–30 First, an oven-dried 500 mL, three-necked flask equipped with a Teflon coated mechanical stir bar, constant-voltage funnel was charged with ferric chloride hexahydrate (21.624 g, 0.08 mol), ferrous chloride tetrahydrate (8.419 g, >0.04 mol) and sodium citrate dehydrate (C6H5Na3O7·2H2O, 0.510 g) and then sealed tightly with a rubber septum. The flask was evacuated and backfilled with nitrogen three times under mechanical stirring. Then, 100 mL of distilled water was added via a dropping funnel under vigorous stirring. The mixture was heated to 60 °C and stirred for 30 min. Finally, 100 mL of aqueous solution containing (25 wt%) ammonium hydroxide 25 mL was added drop-wise to the above mixture, and it immediately turned black. The reaction mixture was heated to 80 °C and kept for 2 h and then allowed to cool to room temperature. The black products were obtained with the help of a magnet and washed with distilled water, ethanol 3 times respectively and then dried under vacuum at 50 °C.2.4 Synthesis of aminopropyl modified silica coated Fe3O4 magnetic nanoparticles (Fe3O4@SiO2–NH2)
Silica coated Fe3O4 magnetic nanoparticles (Fe3O4@SiO2) was synthesized according to the procedure reported in the previous literatures.28,29,31,32 Chloropropyl modified silica coated Fe3O4 magnetic nanoparticles (Fe3O4@SiO2–Cl) was synthesized according to the procedure reported in the previous literature.33 The obtained Fe3O4@SiO2–Cl (3 g) was suspended in acetonitrile (200 mL) with sonication about 20 min. To the mixture, ethylenediamine (10 mL) was added drop-wise via a dropping funnel under mechanical stirring at nitrogen atmosphere and the reaction mixture was refluxed for 12 h. After cooling to room temperature, the resulted solid (Fe3O4@SiO2–NH2) was separated magnetically and then washed with absolute ethanol several times and dried under vacuum at 50 °C.2.5 General procedure for the synthesis of nitroalkenes
Typically, the reactions were carried out as follows: an oven-dried 25 mL flask was charged with Fe3O4@SiO2–NH2 (0.1 g) and the aldehyde (0.3 mmol) in CH3NO2 (5 mL). Then the mixture was stirred at 80 °C under air. After the reaction was completed, the solid catalyst was separated magnetically, washed with ethyl acetate and dichloromethane 3 times respectively. Combined the organic layer and dried with anhydrous Na2SO4, then the Na2SO4 was filtered off. After removal of the solvent under reduced pressure, the crude product was purified by preparative TLC to give the pure nitroalkene.2.6 Separation of the catalyst and recycling tests
After completion of the reaction, the solid catalyst was separated from the reaction mixture with the help of a magnet and washed with ethyl acetate and dichloromethane 3 times respectively. Without drying, the recovered catalyst was directly used in next run with new portions of reactants.3 Results and discussion
3.1 Synthesis and characterization of Fe3O4@SiO2–NH2
Scheme 1 shows the sequence of events in the functionalization of Fe3O4 MNPs with ethylenediamine. Firstly, the Fe3O4 MNPs were prepared using reported chemical coprecipitation methods with slight modification. Secondly, the surface of Fe3O4 MNPs was coated with a silica shell by the hydrolysis and condensation of tetraethyl orthosilicate (TEOS) in ethanol/ammonia mixture to yield Fe3O4@SiO2. Then treatment of Fe3O4@SiO2 with chloropropyl triethoxysilane obtained chloropropyl modified Fe3O4@SiO2 (Fe3O4@SiO2–Cl). Finally, Fe3O4@SiO2–NH2 was synthesized via the substitution of Fe3O4@SiO2–Cl with excess of ethylenediamine. The content of nitrogen was determined to be 1.27 mmol g−1 by elemental analysis.FTIR spectra of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2–Cl and Fe3O4@SiO2–NH2 were shown in Fig. 1. The strong adsorption peak at 581 cm−1 corresponded to the characteristic vibration absorption peak of Fe–O bond. The peak at 3386 cm−1 was assigned to the –OH on the surface Fe3O4 particles. The adsorption peaks located at 1613 and 1398 cm−1 can be attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O, which indicates the presence of carboxyl groups on the surface of Fe3O4 NPs. The new absorption peaks in Fig. 1(b) at 1089, 795 and 465 cm−1 were assigned to the Si–O–Si, the peaks at 960 and 3736 cm−1 were assigned to Si–OH moieties. These indicate that the silica has been successfully coated on the surface of Fe3O4 NPs. In Fig. 1(c) and (d) the adsorption peaks at 1459 cm−1 and 2927 cm−1 were related to C–H stretching modes of the propyl groups. The absorption peaks at 3421 cm−1 and 1508–1551 cm−1 were assigned to the stretching vibration and bending vibration of N–H. These suggested that the Fe3O4@SiO2–NH2 was synthesized successfully.
O and C–O, which indicates the presence of carboxyl groups on the surface of Fe3O4 NPs. The new absorption peaks in Fig. 1(b) at 1089, 795 and 465 cm−1 were assigned to the Si–O–Si, the peaks at 960 and 3736 cm−1 were assigned to Si–OH moieties. These indicate that the silica has been successfully coated on the surface of Fe3O4 NPs. In Fig. 1(c) and (d) the adsorption peaks at 1459 cm−1 and 2927 cm−1 were related to C–H stretching modes of the propyl groups. The absorption peaks at 3421 cm−1 and 1508–1551 cm−1 were assigned to the stretching vibration and bending vibration of N–H. These suggested that the Fe3O4@SiO2–NH2 was synthesized successfully.
 | ||
| Fig. 1 FT-IR spectra of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2–Cl (c) and Fe3O4@SiO2–NH2 (d). | ||
The crystalline structures of the Fe3O4 particles, Fe3O4@SiO2 and Fe3O4@SiO2–NH2 were characterized by XRD and the results are shown in Fig. 2. The experimentally obtained patterns were compared with the standard Fe3O4 pattern (JCPDS (The Joint Committee on Powder Diffraction Standards) card no. 19-0629). The diffraction peaks at 2θ = 30.2°, 35.4°, 43.4°, 53.1°, 57.2° and 62.6° correspond to the (220), (311), (400), (422), (511) and (440) Fe3O4 lattice indices respectively.28–31 These results prove that the Fe3O4 obtained has highly crystalline cubic spinel structure. The XRD patterns of Fe3O4@SiO2 (b) and Fe3O4@SiO2–NH2 (c) show an obvious diffusion peak at about 15.3° which is due to the effect of amorphous silica and organic groups.28,31,34,35 These results indicated that the surface of Fe3O4 was coated with a silica shell successfully and Fe3O4@SiO2 was successfully modified by organic groups.
 | ||
| Fig. 2 XRD patterns of Fe3O4 (a), Fe3O4@SiO2 (b) and Fe3O4@SiO2–NH2 (c). | ||
The surface morphology and size of Fe3O4 (a) and Fe3O4@SiO2 (b) were observed by transmission electron microscopy (TEM) as shown in Fig. 3.
 | ||
| Fig. 3 TEM images of Fe3O4 (a) and Fe3O4@SiO2 (b). | ||
It can be seen that the Fe3O4 particles are successfully packaged by silica. Although the presence of multinuclei Fe3O4 cores embedded in SiO2 shell, this will not affect its application as a support in catalysis.3
Thermogravimetric analysis was performed to evaluate the stability of Fe3O4@SiO2–NH2, since the following reaction required heating in an aerobic condition. Fig. 4 indicated the TG spectra of the fresh and spent catalyst recorded up to 1000 °C. As shown in Fig. 4, when Fe3O4@SiO2–NH2 was heated from room temperature to 80 °C, the loss of weight was only 0.7%, which proved that Fe3O4@SiO2–NH2 was relatively stable as a catalyst in experimental conditions and it may be attributed to the bound water. Also, the decomposition of the surface organic groups of catalyst may contribute to the loss part as the small amount of nitrogen leaching occurred in recycled catalyst.
 | ||
| Fig. 4 TGA spectra of the fresh (a) and spent (b) Fe3O4@SiO2–NH2. | ||
X-ray photoelectron spectrum of Fe3O4@SiO2–NH2 was shown in Fig. 5. The O 1s peak at 536.0 eV and Si 2p peak at 106.4 eV corresponded to the SiO2-type material. The C 1s peak at 284.7 eV and the N 1s peak at 399.6 eV confirmed that ethylenediamine was successfully grafted to the surface of Fe3O4@SiO2.33
 | ||
| Fig. 5 XPS spectrum of Fe3O4@SiO2–NH2. | ||
3.2 Cooperative catalysis for selective synthesis of nitroalkenes
 | ||
| Scheme 2 Selective synthesis of nitroalkene in the presence of Fe3O4@SiO2–NH2. | ||
| Entrya | Amount of catalyst (g) | Time (h) | Temperature (°C) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: anisaldehyde (0.3 mmol) and nitromethane (5.0 mL), in air.b Isolated yield based on aldehyde. | ||||
| 1 | 0.050 | 4 | 80 | 55 |
| 2 | 0.075 | 4 | 80 | 68 |
| 3 | 0.100 | 4 | 80 | 97 |
| 4 | 0.125 | 4 | 80 | 98 |
| 5 | 0.150 | 4 | 80 | >99 |
| 6 | 0.100 | 4 | 30 | Trace |
| 7 | 0.100 | 4 | 60 | 69 |
| 8 | 0.100 | 2 | 80 | 65 |
| 9 | 0.100 | 3 | 80 | 87 |
Under the optimal conditions in Table 1, a range of representative aromatic aldehydes were applied to evaluate the scope of the reaction as shown in Table 2. The Fe3O4@SiO2–NH2 catalyst was effective and selective toward most of the substrates. Both aldehydes bearing electron-rich and electron-withdrawing substituents on the aromatic rings reacting with nitromethane proceeded well and selectively gave the corresponding nitroalkenes in a moderate or excellent yield. However, the electron-withdrawing substituted aromatic aldehydes were slightly inferior to the electron-rich substituted aromatic aldehydes even under prolonged reaction times. As expected, the reaction between nitromethane and heterocyclic aromatic aldehyde was in a moderate yield.
| Entrya | Aldehyde | Nitroalkene | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Unless otherwise specified, all the reactions were carried out using aromatic aldehydes or heterocyclic aromatic aldehyde (0.3 mmol), nitromethane (5.0 mL), Fe3O4@SiO2–NH2 (0.100 g), under 80 °C in air.b Isolated yield based on aldehyde. | ||||
| 1 |  |
 |
4 | 97 |
| 2 |  |
 |
4 | 95 |
| 3 |  |
 |
4 | 88 |
| 4 | 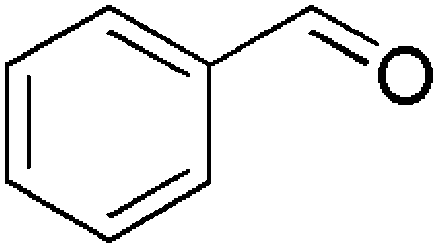 |
 |
4 | 97 |
| 5 |  |
 |
6 | 74 |
| 6 | 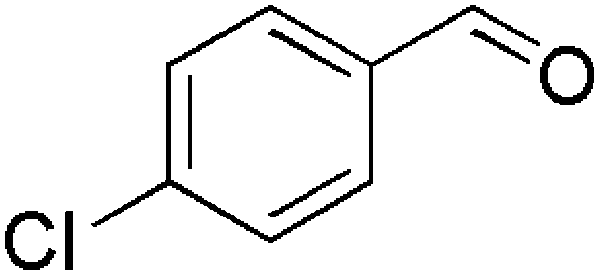 |
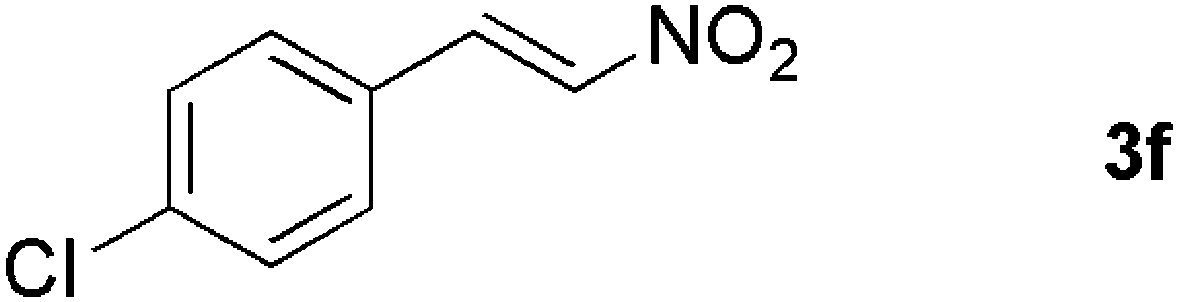 |
6 | 87 |
| 7 |  |
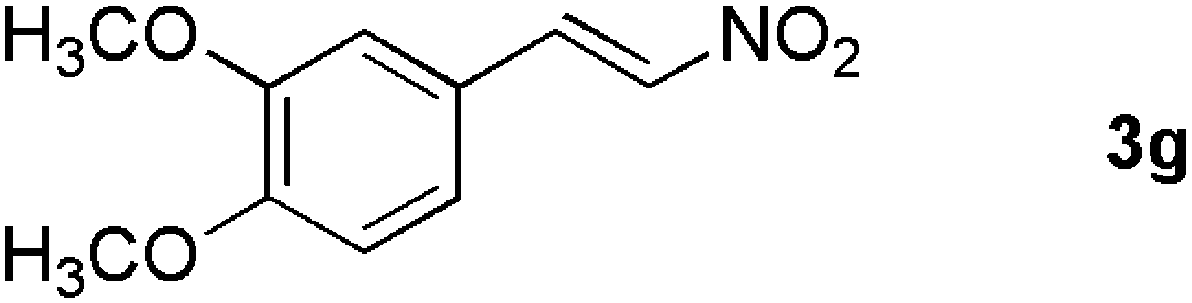 |
4 | 93 |
| 8 |  |
 |
4 | 89 |
| 9 |  |
 |
4 | 56 |
| 10 |  |
 |
4 | 97 |
In order to confirm the possible cooperative trifunctional catalysis mechanism, the following experiments were carried out as shown in Table 3. The content of primary amine nitrogen in Fe3O4@SiO2–NH2 (0.200 g) and NH2CH2CH2NH2 (0.004 g) are equal. When ethylenediamine (0.004 g) was used in catalytic reaction, the yield was 63% (Table 3, entry 1). In a similar way, when using diethylamine (0.010 g) for catalytic reaction, the yield was only 47% (Table 3, entry 2). When ethylenediamine (0.004 g) and diethylamine (0.010 g) were used in catalytic reaction together, the yield was up to 70% (Table 3, entry 3). These indicated that primary amine and secondary amine collaboratively accelerated the reaction. In order to eliminate the effect of chlorine substituent and Si–OH groups, Fe3O4@SiO2–Cl (0.200 g) was used for the reaction between anisaldehyde and nitromethane (Table 3, entry 4). Results showed that the separated chlorine substituent or Si–OH groups could hardly catalyze the reaction. When using Fe3O4@SiO2–NH2 (0.200 g) to catalyze the reaction, the yield was high to 95% (Table 3, entry 5). These results suggested that the cooperative catalysis of primary and secondary amines was enhanced by Si–OH groups on the surface of Fe3O4@SiO2–NH2. In addition, the yield of 1,3-dinitroalkanes compounds 4a could be obtained in 26% by prolonged reaction time to 8 h. Combined with the experimental data and literatures,17–19,21,22,25–27 we speculated that the reaction between nitromethane and aromatic aldehyde under Fe3O4@SiO2–NH2 was through a trifunctional cooperative catalysis mechanism in Scheme 3.
| Entrya | Catalyst (g) | Time (h) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: anisaldehyde (0.6 mmol) and nitromethane (5.0 mL), under 80 °C in air.b Isolated yield based on anisaldehyde.c Yield of 1,3-dinitroalkanes compounds. | |||
| 1 | NH2CH2CH2NH2 (0.004 g) | 4 | 63 |
| 2 | NH(CH2CH3)2 (0.010 g) | 4 | 47 |
| 3 | NH2CH2CH2NH2 (0.004 g) + NH(CH2CH3)2 (0.010 g) | 4 | 70 |
| 4 | Fe3O4@SiO2@Cl (0.200 g) | 4 | Trace |
| 5 | Fe3O4@SiO2@NH2 (0.200 g) | 4 | 95 |
| 6 | Fe3O4@SiO2@NH2 (0.200 g) | 8 | 71 (26)c |
 | ||
| Scheme 3 A possible cooperative trifunctional catalysis mechanism. | ||
The aromatic aldehyde 1a is activated by both the silanol and primary amine on the surface of the Fe3O4@SiO2–NH2 catalyst and reacts with an amino group to form an imine intermediate, the α-proton of nitromethane 2a is abstracted by secondary amine or another amino group, accompanied by the nucleophilic attack of the deprotonated nitromethane on imine intermediate, which is also assisted by the silanol group resulting in the product nitroalkenes 3a. Additionally, the Michael reaction between nitromethane 2a which is activated by secondary amine or another amino group and nitroalkenes 3a which might also be activated by the surface silanol group possibly occurs to obtain the 1,3-dinitroalkanes compound 4a. Nucleophilic attack of the deprotonated nitromethane on aromatic aldehyde with electron-withdrawing groups on the aromatic rings might easily occur without the formation of imine intermediates, resulting in lower selectivity to nitroalkene formation.27
4 Conclusions
In conclusion, a magnetically separable trifunctional nanocatalyst Fe3O4@SiO2–NH2 was designed and synthesized. The catalyst was found to be highly active for selective synthesis of nitroalkenes with nitromethane and aromatic aldehyde involving cooperative catalysis of primary amine, secondary amine and Si–OH groups. Under the optimized conditions, various representative substrates were extended to obtain the corresponding product in a moderate or excellent yield. In addition, a possible cooperative trifunctional catalysis mechanism was also proposed. The magnetic trifunctional nanocatalyst Fe3O4@SiO2–NH2 has the prominent properties as follows: (a) simple and efficient separation (b) cooperatively accelerate a single reaction. The molecular design of more multifunctional surface and further applications to more organic reaction are currently being investigated in our laboratory.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 20972109) and National Youth Science Foundation of China (No. 51403151).References
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- X. X. Wang, P. B. Hu, F. J. Xue and Y. P. Wei, Carbohydr. Polym., 2014, 114, 476–483 CrossRef CAS PubMed.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- W. Teunissen, A. A. Bol and J. W. Geus, Catal. Today, 1999, 48, 329–336 CrossRef CAS.
- M. B. Gawande, P. S. Brancoa and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 Search PubMed.
- O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1887–1894 Search PubMed.
- N. Takenaka, J. Chen, B. Captain, R. S. Sarangthem and A. Chandrakumar, J. Am. Chem. Soc., 2010, 132, 4536–4537 CrossRef CAS PubMed.
- J. Wu, X. Li, F. Wu and B. Wan, Org. Lett., 2011, 13, 4834–4837 CrossRef CAS PubMed.
- J. W. Xie, Z. Wang, W. J. Yang, L. C. Kong and D. C. Xu, Org. Biomol. Chem., 2009, 7, 4352–4354 CAS.
- Y. K. Liu, H. Liu, W. Du, L. Yue and Y. C. Chen, Chem.–Eur. J., 2008, 14, 9873–9877 CrossRef CAS PubMed.
- D. Enders, M. R. M. Hüttl, J. Runsink, G. Raabe and B. Wendt, Angew. Chem., Int. Ed., 2007, 46, 467–469 CrossRef CAS PubMed.
- L. Celano, C. Carabio, R. Frache, N. Cataldo, H. Cerecetto, M. González and L. Thomson, Eur. J. Med. Chem., 2014, 74, 31–40 CrossRef CAS PubMed.
- S. P. Chavan, S. Garai and K. P. Pawar, Tetrahedron Lett., 2013, 54, 2137–2139 CrossRef CAS PubMed.
- S. Lühr, M. Vilches-Herrera, A. Fierro, R. R. Ramsay, D. E. Edmondson, M. Reyes-Parada, B. K. Cassels and P. Iturriaga-Vásquez, Bioorg. Med. Chem., 2010, 18, 1388–1395 CrossRef PubMed.
- H. X. Yu, J. W. Xie, Y. J. Zhong, F. M. Zhang and W. D. Zhu, Catal. Commun., 2012, 29, 101–104 CrossRef CAS PubMed.
- S. Jalal, S. Sarkar, K. Bera, S. Maiti and U. Jana, Eur. J. Org. Chem., 2013, 4823–4828 CrossRef CAS PubMed.
- G. Demicheli, R. Maggi, A. Mazzacani, P. Righi, G. Sartori and F. Bigi, Tetrahedron Lett., 2001, 42, 2401–2403 CrossRef CAS.
- H. Hagiwara, M. Sekifuji, N. Tsubokawa, T. Hoshi and T. Suzuki, Chem. Lett., 2009, 38, 790–791 CrossRef CAS.
- S. Shylesh, A. Wagner, A. Seifert, S. Ernst and W. R. Thiel, Chem.–Eur. J., 2009, 15, 7052–7062 CrossRef CAS PubMed.
- S. M. Ribeiro, A. C. Serra and A. M. R. Gonsalves, Appl. Catal., A, 2011, 399, 126–133 CrossRef CAS PubMed.
- G. Sartori, F. Bigi, R. Maggi, R. Sartorio, D. J. Macquarrie, M. Lenarda, L. Storaro, S. Coluccia and G. Martra, J. Catal., 2004, 222, 410–418 CrossRef CAS PubMed.
- J. X. Yang, J. Dong, X. Lü, Q. Zhang, W. Ding and X. X. Shi, Chin. J. Chem., 2012, 30, 2827–2833 CrossRef CAS PubMed.
- A. E. Kadib, K. Molvinger, M. Bousmina and D. Brunel, J. Catal., 2010, 273, 147–155 CrossRef PubMed.
- A. Alizadeh, M. M. Khodaei and A. Eshghi, J. Org. Chem., 2010, 75, 8295–8298 CrossRef CAS PubMed.
- K. Motokura, M. Tada and Y. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 9230–9235 CrossRef CAS PubMed.
- L. Sun, S. C. Hu, H. M. Sun, H. L. Guo, H. D. Zhu, M. X. Liu and H. H. Sun, RSC Adv., 2015, 5, 11837–11844 RSC.
- A. Bayat, M. Shakourian-Fard, N. Ehyaei and M. M. Hashemi, RSC Adv., 2015, 5, 22503–22509 RSC.
- Y. L. Zhang, W. W. Yan, Z. M. Sun, X. C. Li and J. P. Gao, RSC Adv., 2014, 4, 38040–38047 RSC.
- F. Liu, F. G. Niu, N. Peng, Y. J. Sun and Y. J. Yang, RSC Adv., 2015, 5, 18128–18136 RSC.
- Y. H. Deng, Y. Cai, Z. K. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Y. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- L. Chen, B. D. Li and D. B. Liu, Catal. Lett., 2014, 144, 1053–1061 CrossRef CAS.
- M. Esmaeilpour, A. R. Sardarian and J. Javidi, J. Organomet. Chem., 2014, 749, 233–240 CrossRef CAS PubMed.
- M. Esmaeilpour, A. R. Sardarian and J. Javidi, Appl. Catal., A, 2012, 445–446, 359–367 CrossRef CAS PubMed.
- D. Kühbeck, G. Saidulu, K. R. Reddy and D. D. Díaz, Green Chem., 2012, 14, 378–392 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11798d |
| This journal is © The Royal Society of Chemistry 2015 |