
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High performance alkyd resins synthesized from postconsumer PET bottles
P. M.
Spasojević
*a,
V. V.
Panić
a,
J. V.
Džunuzović
b,
A. D.
Marinković
c,
A. J. J.
Woortman
d,
K.
Loos
d and
I. G.
Popović
c
aInnovation Center of Faculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, Belgrade 11120, Serbia. E-mail: pspasojevic@tmf.bg.ac.rs
bInstitute of Chemistry, Technology and Metallurgy (ICTM) – Center of Chemistry, University of Belgrade, Studentski trg 12-16, 11000 Belgrade, Serbia
cFaculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, 11120 Belgrade, Serbia
dDepartment of Polymer Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 13th July 2015
Abstract
The glycolytic recycling of waste PET presents a challenge for the production of secondary value-added products, such as alkyd resins. A way to overcome the unsatisfactory mechanical, drying and chemical resistance properties of alkyds obtained from difunctional glycolyzates was proposed. Waste PET was glycolyzed using multifunctional alcohols: glycerol (G), trimethylolethane (TME), trimethylolpropane (TMP) and pentaerythritol (PE), giving tetra- and hexa-functional glycolyzates and, for comparison, using diethylene glycol (DEG), propylene glycol (PG) and dipropylene glycol (DPG) giving di-functional glycolyzates. The obtained glycolyzates were examined by 1H and 13C NMR, FTIR spectroscopy and elemental analysis and further used in the synthesis of alkyd resins. The properties of the prepared alkyd resins (acid, hydroxyl and iodine values, color, average molar masses and molar mass distributions, viscosity, drying time, hardness, flexibility, gloss, adhesion and chemical resistance) were investigated with respect to the functionality and the structure of the used glycolyzates. Alkyd resins derived from multifunctional glycolyzates (TME and TMP) showed considerably enhanced properties compared to those produced from difunctional glycolyzates and also to conventional general purpose resins.
Introduction
Alkyd resins were first synthesized in the 1920s and, due to their very good performances, have persisted in the endless competition with the growing synthetic-based polymer resins till now. Because of the great compatibility with many polymers and the large number of formulations, alkyds have proven to be superior to other systems in many applications with special demands.1 This versatility, combined with their biodegradability and the fact that the raw materials used in the synthesis, except for phthalic anhydride, are from renewable sources – biological and possibly recyclable, makes alkyds very attractive both from the environmental and the economical point of view.2Polyethylene terephthalate (PET) is thermoplastic polyester of excellent tensile strength, transparency, chemical resistance and safety, processability and reasonable thermal stability. Due to its characteristic features, PET has become indispensable in the field of beverage containers. PET bottles have become very popular because they are light for on-the-go consumption, resealable, easy to handle and do not break. Nowadays, PET bottles are used for mineral water, carbonated soft drinks, energy drinks, ready-to drink teas, as well as for more sensitive beverages like beer, wine and juices.3 PET packaging is also increasingly making inroads into the markets of other end-use sectors, such as packaged food, household cleaning products and pharmaceuticals.
In 2013 the global consumption of PET packaging was 15.4 Mt.4 The newest report from an ongoing series of the packaging market predicts that the global consumption of PET packaging will grow to over 20 Mt by 2019 – a 4.6% increase per annum during the period 2014–2019.5
Although PET does not have any side effects on the human organism and does not create a direct hazard to the environment, due to such widespread use (about 18% of the world polymer production), the inability to decompose in nature and the fact that the life time of most PET waste is very short, waste PET is seen as a harmful material. Since every year huge amounts of PET waste reach the ultimate consumers, such a trend has created a serious environmental problem and PET now accounts for more than 8% by weight and 12% by volume of the world's solid waste.6 The urge to deal with this type of pollution, increased by the fact that its disposing to landfill is becoming undesirable due to rising costs, legislation pressures and the poor biodegradability, provides a huge potential for the development of different technologies for the successful recycling of waste PET.7,8
Nowadays, PET is considered as one of the easiest materials to recycle, because essentially, all basic recycling procedures could be applied for its recycling. There are four approaches: primary or in-plant recycling of the scrap material of controlled history, secondary or mechanical, tertiary or chemical and quaternary or recovery of energy through incineration. However, the only acceptable one according to the principles of sustainable development is chemical recycling that leads back to the formation of PET original monomer forms or the generation of various value-added products.9
One of the most investigated chemical ways for PET recycling, but still with great potential for research, is glycolytic depolymerization (glycolysis). This process involves the solvolytic cleavage of PET chains by using diverse glycols to give the so called glycolyzates consisted of monomers for the repolymerization of PET (bis(hydroxyalkyl)terephthalate) and/or oligomers. Ethylene glycol (EG) is the most usually used solvent in PET glycolysis,10–12 but also diethylene glycol (DEG),13 triethylene glycol (TEG),14 propylene glycol (PG)15 or dipropylene glycol (DPG).16 Research efforts have been aimed in three directions. One group of authors has dealt with the optimization of the reaction conditions; others have sought more eco-friendly glycolytic processes, while the third group has investigated the possibilities for applications of the glycolysis product without the separation of oligomers.17 The latter showed that glycolyzates can be used as building blocks in the synthesis of other polymers with higher economical values, mainly unsaturated polyesters, but also polyurethane foams, copolyesters, polyurethane coatings, low-temperature and UV curable resins, alkyd resins, etc.18
The use of PET glycolyzates in the synthesis of alkyd resins involves their employment as a hydroxyl component instead of a certain amount of diverse glycols. Most of the reported PET glycolyzates for the synthesis of alkyd resins were difunctional.19,20 Difunctional components provide lower branching and, thus, lower molar masses of alkyd resins and, consequently, insufficient mechanical, drying, chemical resistance properties, etc.21,22
In order to overcome the reported drawbacks of alkyds, we used multifunctional glycolyzates to prepare alkyd resins with improved applicative properties. For that purpose waste PET was glycolyzed using four multifunctional alcohols: glycerol (G), trimethylolethane (TME), trimethylolpropane (TMP) and pentaerythritol (PE), giving tetra- and hexa-functional glycolyzates. For comparison, difunctional PET glycolyzates were also synthesized using diethylene glycol, propylene glycol and dipropylene glycol.
The applied process was further justified because two important environmental issues were resolved: (1) the use of waste material promoted the reduction of the amount of waste PET disposed to landfills or just dumped, and (2) the replacement of a certain amount of alcohols with the derived PET glycolyzates also reduced the amount of phthalic anhydride in the feed composition, because PET glycolyzate also introduced an aromatic ring.
Experimental
Materials
PET flakes were obtained by the grinding of postconsumer PET clear and colorless bottles. The ground pieces (approximately 10–20 mesh) were washed with acetone and dried at 110 °C for 4 h. Linseed oil fatty acid (LFA), used in the preparation of the alkyd resins, was kindly supplied by Croda GmbH, Nettetal, Germany. Diethylene glycol, propylene glycol, dipropylene glycol, trimethylolethane, trimethylolpropane, glycerol, pentaerythritol, phthalic anhydride (PA), tetrabutyl titanate (TBT), xylene and mineral spirits were obtained from Sigma-Aldrich and were used without any further purification.PET glycolysis
The glycolysis of waste PET was carried out in a 500 mL four-necked flask equipped with a thermometer, nitrogen inlet tube, a reflux condenser and a mechanical stirrer. Seven alcohols of different functionality were used for PET recycling: diethylene glycol, propylene glycol, dipropylene glycol, trimethylolethane, trimethylolpropane, glycerol and pentaerythritol. In all experiments 96 g of PET, equivalent to 0.48 mol of the repeating unit in the PET chain, 1.44 mol of alcohol and 0.28 mL of TBT were charged to the reactor. The reaction mixture was heated at 210 °C under nitrogen atmosphere for 6 h. At the beginning, the reaction mixture was heterogeneous (solid PET and liquid alcohols) but, after a 3–4 hours the solid phase was no longer observed and the reaction continued in one single liquid phase. After completion of the glycolysis, all reaction mixtures, except the one with the PE, were dissolved in dichloromethane and washed two times with 150 mL of distilled water in order to remove excess of alcohols. Afterwards, the mixtures were filtered with a sintered glass filter (Borosil Glass Works Ltd, porosity grade 3) and dried at 80 °C for 12 hours. The purification of the product obtained by the use of PE was firstly performed by dissolution in methanol at 50 °C. The next step involved pouring of the obtained viscous solution into distilled water, keeping 50 °C during the addition. The efficient mixing of the obtained emulsion was necessary (using a mechanical stirrer (IKA)), over the whole period of addition and thereafter during slow temperature decrease (without external cooling) of the obtained two-phase system. Approaching room temperature, the rate of stirring was proportionally decreased, providing separation of the upper aqueous phase, which contained non-reacted PE. The hydroxyl numbers of the glycolyzed products were determined afterwards by the reported analytical method.23 The obtained glycolyzates were denoted as g-alcohol abbreviation.Synthesis of alkyd resins
Seven different alkyds, having oil contents of around 48%, were synthesized using alcohols obtained by waste PET glycolysis. The amounts of reactants were calculated using a method based on the alkyd constant value.24 The excess of hydroxyl groups was around 20% for all synthesized alkyds. The feed compositions are shown in Table 1. All alkyd resins were prepared in a four-necked reactor equipped with a stirrer, thermometer, nitrogen inlet tube and Dean–Stark separator. Xylene was used as the azeotropic solvent. All ingredients were charged into the reactor and the reaction mixture was heated to 220 °C. The acid value of the reaction mixture was measured by taking sample aliquots at various time intervals. The reaction was quenched by allowing it to cool when the acid value was about 9 mg KOH per g. The alkyd resins were diluted to 60% of solid content by adding xylene. A 0.2 wt% mixture of calcium, zirconium and manganese salts was used as a drying catalyst in all experiments.| wt% | AR-DEG | AR-PG | AR-DPG | AR-G | AR-TME | AR-TMP | AR-PE |
|---|---|---|---|---|---|---|---|
| g-DEG | 20.2 | — | — | — | — | — | — |
| g-PG | — | 20.2 | — | — | — | — | — |
| g-DPG | — | — | 21.2 | — | — | — | — |
| g-G | — | — | — | 18.4 | — | — | — |
| g-TME | — | — | — | — | 19.9 | — | — |
| g-TMP | — | — | — | — | — | 19.6 | — |
| g-PE | — | — | — | — | — | — | 20.5 |
| LFA | 43.4 | 40.3 | 43.4 | 45.9 | 44.7 | 44.1 | 44 |
| PA | 22.2 | 25.3 | 22.2 | 21.5 | 21.4 | 22.6 | 21.5 |
| PE | 7.1 | 7.1 | 7.1 | 7.1 | 7.0 | 5.9 | 7.0 |
| TMP | 7.1 | 7.1 | 6.1 | 7.1 | 7.0 | 7.8 | 7.0 |
As mentioned, xylene was employed as an azeotropic solvent in the alkyds synthesis and as diluents for alkyd resins. In favor of the use of this solvent stands the fact that waterborne alkyd resins do not perform as well and are more expensive, despite having lower solids contents. Their dry time is also affected by temperature and humidity and their storage stability is usually lower than solvent-borne alkyds.25 Moreover, although xylene is currently produced mostly by the catalytic reforming of various crude oil streams, it has been shown that it can be produced from biomass, which contains six-carbon (glucose) and five-carbon (e.g., xylose) sugars.26 The high yields (90%) and selectivity (90%) and renewable nature of the proposed processes27 makes xylene acceptable solvent for green production of alkyd resins, until some better green solvent or waterborne formulation is found.
Characterization of the products of glycolysis and synthesized alkyd resins
The hydroxyl and the acid values of the synthesized alkyd resins were determined by standard methods.23,28 Thin films of alkyd resins were deposited on KBr discs and Fourier-transform infrared (FTIR) spectra were recorded using a FTIR BOMEM MB 100 instrument. The 1H and 13C NMR spectral measurements of the alkyd resins were performed on a Varian Gemini 2000 (200 MHz). The spectra were recorded at room temperature in deuterated chloroform (CDCl3). The chemical shifts were expressed in ppm values referenced to tetramethylsilane (TMS). The viscosity of the alkyd resins was determined by using a Brookfield RV/DV-II type rotational viscometer at 25 °C. The number and mass average molar masses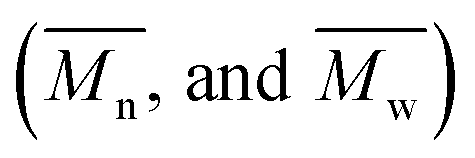 and polydispersity index (PDI) were measured by gel permeation chromatography (GPC) using a Viscotek GPC equipped with three detectors (LS detector: Viscotek Ralls detector; VS detector: Viscotek Viscometer Model H502; RI detector: Shodex RI-71 Refractive Index detector), using a guard column (PLgel 5 μm Guard, 50 mm) and two columns (PLgel 5 μm MIXED-C, 300 mm, from Agilent Technologies) at 30 °C. High performance liquid chromatography grade chloroform was used as the eluent at a flow rate of 0.8 mL min−1. Low polydispersity linear polystyrene standards (Agilent and Polymer Laboratories) were used to construct a calibration curve based on universal GPC calibration.
and polydispersity index (PDI) were measured by gel permeation chromatography (GPC) using a Viscotek GPC equipped with three detectors (LS detector: Viscotek Ralls detector; VS detector: Viscotek Viscometer Model H502; RI detector: Shodex RI-71 Refractive Index detector), using a guard column (PLgel 5 μm Guard, 50 mm) and two columns (PLgel 5 μm MIXED-C, 300 mm, from Agilent Technologies) at 30 °C. High performance liquid chromatography grade chloroform was used as the eluent at a flow rate of 0.8 mL min−1. Low polydispersity linear polystyrene standards (Agilent and Polymer Laboratories) were used to construct a calibration curve based on universal GPC calibration.
Dry film properties
The alkyd resins were applied by a wire-wound applicator (wet thickness of 100 μm) on mild steel 15 cm × 10 cm panels for evaluating flexibility, gloss and adhesion and 10 cm × 10 cm glass panels for evaluating hardness, water, alkali, acid and solvent resistances. The coatings were left to dry for 21 days at room temperature prior to the measurements. The flexibility was measured according to the ISO 1520 using a cupping tester (Byk PF-5400, BYK-Gardner GmbH, Geretsried, Germany). The adhesion of the films to the substrate was characterized using the crosshatch adhesion method (ASTM D 3359), which is a simple and effective method for evaluating adhesion. Gloss was measured by a single angle gloss meter (AG-4442, BYK-Gardner GmbH, Geretsried, Germany). All measurements were done at an angle of 60° (ASTM D 523). Hardness was measured by a pendulum hardness tester (Byk PH-5858, BYK-Gardner GmbH, Geretsried, Germany) using a König pendulum according to ASTM D 4366. The thermal properties of the coatings were investigated by differential scanning calorimetry (DSC) using a Perkin Elmer DSC-2 thermal analyzer under nitrogen (flow rate 26 mL min−1). The 10–20 mg of coating sample was heated from −60 °C to 100 °C and the heating rate was set to 20 °C min−1. In order to erase the thermal history of the sample, all calculations were done using the data acquired from the second cycle. The coating resistance to distilled water, NaCl, NaOH, H2SO4, acetone and xylene was investigated as follows. A piece of absorbent gauze (2 cm × 2 cm) swollen in a solvent (distilled water, NaCl solution) was put on a film-coated glass panel. The panel was covered with a laboratory dish and kept at 20 ± 2 °C for 24 h. The conditions of the films were visually observed.29Results and discussion
The aim of this work was to obtain alkyd resins from waste PET and to overcome the drawbacks of alkyds prepared from waste PET glycolyzates reported in the literature. In this context we have tried to prepare multifunctional alcohols through the reaction of waste PET glycolysis and to use them for the synthesis of alkyd resins.Glycolysis of PET
The reactions of PET waste glycolysis with DEG, PG, DPG, G, TME, TMP and PE were performed in the presence of TBT catalysts. The obtained glycolyzates were characterized by determination of the hydroxyl (HV) and acid (AV) values, and elemental, as well as FTIR and 1H and 13C NMR analysis. The HV and AV values and the data obtained from elemental analysis of the glycolyzed products are presented in Table 2.| Glycolyzate | HVtheor., mg KOH per g | HVexp., mg KOH per g | AVexp., mg KOH per g | %C | %H | %Oa | |
|---|---|---|---|---|---|---|---|
| a The oxygen percent was calculated as the difference to 100%. | |||||||
| g-DEG | 328 | 317 | 5 | Exp. | 55.93 | 6.63 | 37.34 |
| Calc. | 56.13 | 6.48 | 37.39 | ||||
| g-PG | 397 | 412 | 4 | Exp. | 59.04 | 6.70 | 34.26 |
| Calc. | 59.57 | 6.43 | 34.00 | ||||
| g-DPG | 282 | 265 | 6 | Exp. | 61.23 | 7.65 | 31.12 |
| Calc. | 60.29 | 7.59 | 32.12 | ||||
| g-G | 712 | 675 | 4 | Exp. | 54.45 | 5.34 | 40.21 |
| Calc. | 53.50 | 5.77 | 40.73 | ||||
| g-TME | 605 | 610 | 4 | Exp. | 59.20 | 6.51 | 34.29 |
| Calc. | 58.37 | 7.07 | 34.56 | ||||
| g-TMP | 563 | 551 | 7 | Exp. | 59.45 | 8.09 | 32.46 |
| Calc. | 60.29 | 7.59 | 32.12 | ||||
| g-PE | 837 | 815 | 6 | Exp. | 54.52 | 6.30 | 39.18 |
| Calc. | 53.73 | 6.51 | 39.76 | ||||
The hydroxyl-terminated products of glycolysis were obtained by the trans-esterification reaction of PET, leading to chain fragmentation and the formation of the corresponding ester of terephthalic acid. By comparing the hydroxyl values of the obtained glycolyzed products listed in Table 2, and the ones obtained according to the theoretical calculation for purified products, it could be assumed that the main products of glycolysis were esters of terephthalic acid and the used alcohols. The results of elemental analysis further confirmed this presumption.
The products obtained by PET glycolysis were further characterized by FTIR and NMR spectroscopy. The FTIR spectra of the representative samples of the glycolyzed products are shown in Fig. 1. The broad band, in the spectrum of the glycolyzed products, observed between 3472 cm−1 and 3387 cm−1 was attributed to the OH group stretching vibration. The stretching vibrations, asymmetric and symmetric, of the methyl and methylene groups, appeared in the region 2931–2875 cm−1. The band at 1722 cm−1 was assigned to the stretching vibration of the ester carbonyl group. The main absorption bands between 1273 and 1047 cm−1 originated from the stretching vibrations of the C–O group.
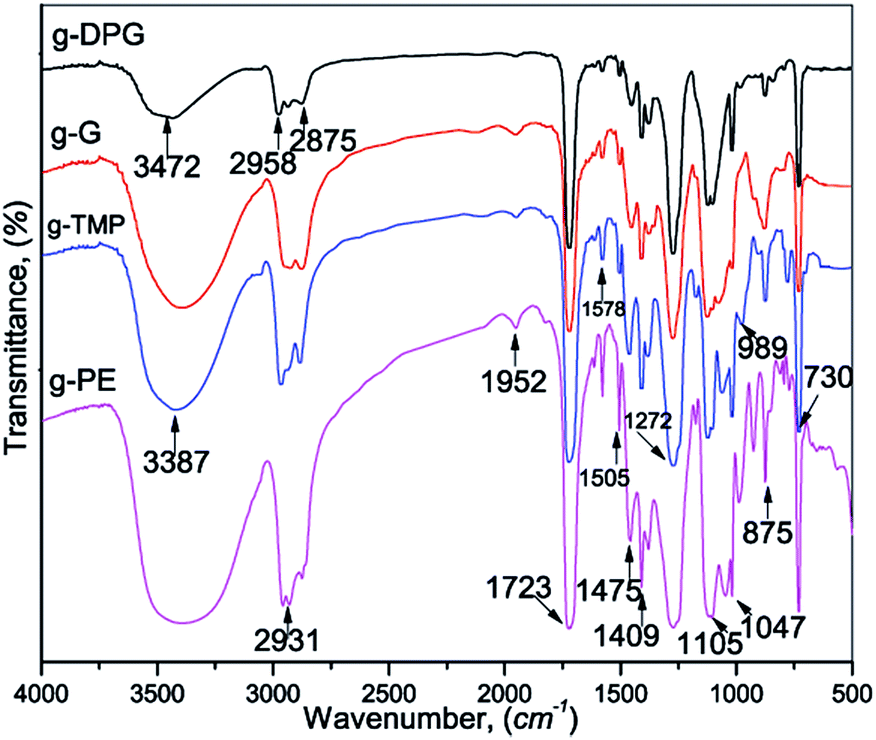 | ||
| Fig. 1 FTIR spectra of the representative samples of the obtained glycolyzed products. | ||
There is some specificity between reacting alcohols: DEG, PG, DPG, G, TME, TMP and PE and the obtained products: g-DEG, g-PG, g-DPG, g-TME, g-TMP and g-PE. Formally three groups of starting alcohols could be recognized: linear molecule with primary hydroxyl (DEG, PG and DPG), branched with primary hydroxyl (TME, TMP and PE) and G is specific alcohol which contains two kinds of hydroxyl groups, both primary and secondary ones. Based on these facts it was very important to consider and discuss effect of hydroxyl group reactivity and structure–properties relationship of obtained AR with respect to structure/reactivity of used alcohol during the trans-esterification and polycondensation reaction.
Results of the 1H and 13C NMR analysis of the glycolyzate are:
g-DPG: 1H NMR: 1.02–1.45 (12H, m, 4 × C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 3.17–3.88 (10H, m, 4 × C2 and 2 × C), 4.10–4.48 (4H, m, 2 × CO2C2), 7.98–8.12 (4H, m, HPh).
3), 3.17–3.88 (10H, m, 4 × C2 and 2 × C), 4.10–4.48 (4H, m, 2 × CO2C2), 7.98–8.12 (4H, m, HPh).
13C NMR: 16.2 and 19.6 (4 × CH3), 66.2–78.4 (4 × CH2 and 4 × CH), 129.6 (4 × CPh), 133.8 (2 × Ph(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) )–COO), 164.9 (2 × COO).
)–COO), 164.9 (2 × COO).
g-TME: 1H NMR: 0.95 (6H, s, 2 × C3); 3.64 (8H, m, 4 × C2OH); 3.66 (4H, m, 4 × CH2O); 4.30–4.55 (8H, m, 4 × COOC2, TME and EG moiety); 7.91–8.14 (4H, m, HPh).
13C NMR: 16.88 (2 × H3); 40.93 ((CH3)(CH2OH)2(CH2)); 67.40 (COOH2, TME moiety); 76.38–77.07 (COOH2, EG moiety); 129.71 (4 × CPh); 133.79 (2 × Ph()COO) 165.81 (2 × OO).
g-TMP: 1H NMR: 0.87 (6H, m, 2 × C3); 3.61 (8H, m, 4 × C2OH); 3.73 (4H, m, 4 × CH2O); 4.28–4.51 (8H, m, 4 × COOC2, TME and EG moiety); 7.20 (2H, m, HPh); 7.85–8.09 (6H, m, HPh).
13C NMR: 7.50 (2 × H3), 22.74 (CH3H2); 43.02 ((CH2CH3)(CH2OH)2(CH2)); 65.16 (COOH2, TME moiety); 76.38–77.65 (COOH2, EG moiety); 129.78 (4 × CPh); 133.79 (2 × Ph()COO); 165.83 (2 × OO).
g-G: 1H NMR: 3.30–3.52 (4H, m, 2 × C2OH), 4.22–4.40 (6H, m, 2 × CH2O, 2 × CHO, 2 × COH), 4.72–4.78 (2H, m, 2 × C2OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 5.07–5.10 (4H, d, 2 × C2OCO), 8.12 (4H, s, CPh).
O), 5.07–5.10 (4H, d, 2 × C2OCO), 8.12 (4H, s, CPh).
13C NMR: 62.80 (2 × H2OH), 66.46 (2 × H2OCO), 69.53 (2 × HOH), 129.83 (4 × CPh), 134.06 (2 × Ph()COO), 165.28 (2 × OO).
Similar results of 1H and 13C NMR of g-TME, g-TMP and g-G was obtained for g-PE.
Characterization of alkyd resins
The alkyd resins were synthesized using the obtained glycolyzates from waste PET as a polyol component. The ARs were characterized by determination of the hydroxyl, acid and iodine values. Also, ARs were thoroughly characterized with respect to the functionality and the structure of the glycolyzate used, by FTIR, NMR, GPC and viscosity analysis. The drying time, hardness, Tg, gloss, flexibility, adhesion and chemical resistance of the dry films were further investigated.The acid value, hydroxyl value, iodine value and color of the synthesized alkyd resins are presented in Table 3.
| Alkyd code | AV, mg KOH per g | HV, mg KOH per g | Iodine value | Color |
|---|---|---|---|---|
| AR-DEG | 9.8 | 22 | 65.3 | Light brown |
| AR-PG | 10.1 | 25 | 59.8 | Pale yellow |
| AR-DPG | 10.0 | 17 | 66.1 | Light brown |
| AR-G | 9.5 | 66 | 59.4 | Brown |
| AR-TME | 8.4 | 47 | 63.4 | Light brown |
| AR-TMP | 7.8 | 43 | 68.2 | Pale yellow |
| AR-PE | 9.2 | 84 | 65.3 | Pale yellow |
It is important to notice that AR-PE has the highest hydroxyl value and that HV of AR-G is higher compared to the value determined for AR-TME and AR-TMP. As presented in Table 3, the HV values are higher for the alkyds obtained from polyfunctional glycolyzates, most likely due to the lower reactivity of the tetra-functional glycolyzate hydroxyl groups (g-PE) in comparison to the tri-functional molecule (g-TME, g-TMP and g-G). On the other hand, hydroxyl groups in di-functional reactants (g-DEG, g-PG and g-DPG) showed the highest conversion degree to ester group. These three types of resins were prepared from different multifunctional glycolyzates whose hydroxyl groups reactivity highly affect the properties of AR based coating. According to the structural analysis, theoretically g-TME and g-TMP would have four primary hydroxyl groups in reaction product if reaction of glycolysis take place at equimolar ratio ester (PET)/hydroxyl group. On the other hand, g-G product would contain two primary and two secondary hydroxyl groups. The secondary hydroxyl groups, present in g-G, are far less reactive than the primary ones, which led to the lower conversion to the ester group and thus higher hydroxyl number of AR-G. The iodine values of all obtained alkyd resins were similar, meaning that the double bonds were stable during synthesis. It could be expected that reaction of all primary hydroxyl groups, present in g-PE, would produce the highest cross-linking density and thus lowest HV value. On the contrary, the highest HV value indicates that when two groups react and form linear chain, reactivity of the third hydroxyl group is highly reduced by sterical repulsion between oligomeric neighboring functionalities of hydroxyl group and attacking molecule containing carboxyl moiety.
As the obtained glycolyzates, the prepared alkyd resins were the broad band, in the spectrum of the representative alkyd resins (Fig. 2), observed at 3520 cm−1 was attributed to the OH group stretching vibration, while the aromatic and vinyl C–H stretching vibration could be observed as a shoulder at ≈3009 cm−1. Overlapping stretching vibrations of the CH3 and CH2 groups, asymmetric and symmetric, were located at 2961, 2927 and 2855 cm−1, respectively, and the corresponding bending vibrations appeared at 1463 and 1385 cm−1, respectively. The stretching vibration of the ester carbonyl group appeared at 1728 cm−1. In the FTIR spectrum of the alkyd resins, the band at 976 cm−1 corresponded to the CC stretching (skeletal) vibration of the benzene ring. The two sharp and moderate bands observed at 740 and 730 cm−1 were assigned to the skeletal deformation, γ(CH), vibration of the benzene ring.
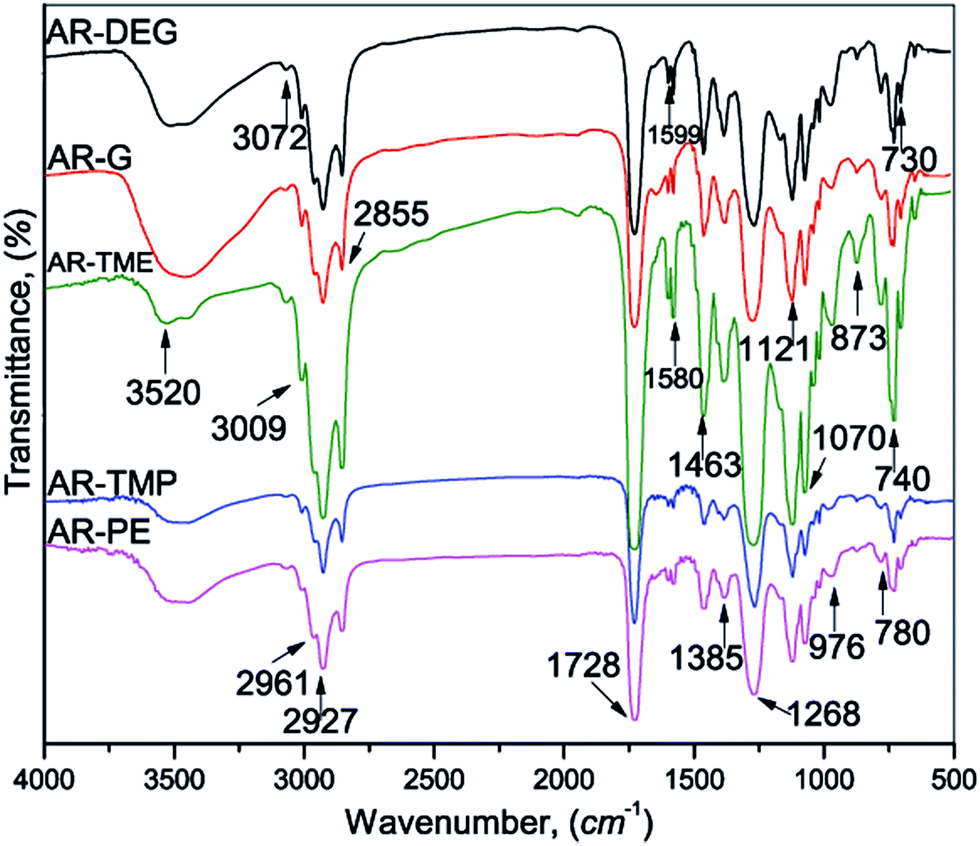 | ||
| Fig. 2 FTIR spectra of the synthesized alkyd resins. | ||
The results of the 1H and 13C NMR analysis of the alkyds synthesized from g-DPG, g-PE and g-G are summarized below:
Results of 1H and 13C NMR analysis of AR-DPG are as follow.
1H NMR: 0.82–1.00 (15H, m, 5 × C3, LFA moiety), 1.26 (70H, m, 35 × C2, LFA moiety), 1.50–1.59 (3H, m, 1 × C3, TMP moiety), 2.03–2.15 (20H, m, 10 × C2–CHCH–, LFA moiety), 2.36 (10H, m, 5 × C2–COO–), 2.78–2.81 (10H, m, 5 × –CHCH–C2–CHCH–, LFA moiety), 3.17–3.88 (10H, m, 4 × C2 and 2 × C, DPG moiety), 4.10–4.48 (4H, m, 2 × COOC2), 4.11–4.40 (10H, m, 5 × C2–, TMP and PE moiety), 5.34–5.36 (20H, m, 10 × –CC–, LFA moiety), 7.75 (2H, m, HPh, PA), 8.08 (m, 7H, HPh).
13C NMR: 14.02 and 18.30 (6 × H3), 22.56–33.99 (H2 carbons in FLA moiety), 60.67–62.27, 76.4–77.6 (H2 carbons in DPG moiety), 63.85 (H2 carbons in TMP and PE moiety), 127.09–128.45 (8 × CPh), 131.7 (2 × Ph()–COO, PA moiety), 132.8 (4 × Ph()–COO), 167.29 (O–Ph–O), 173.1 (O–Ph–O, PA moiety) (Fig. 3).
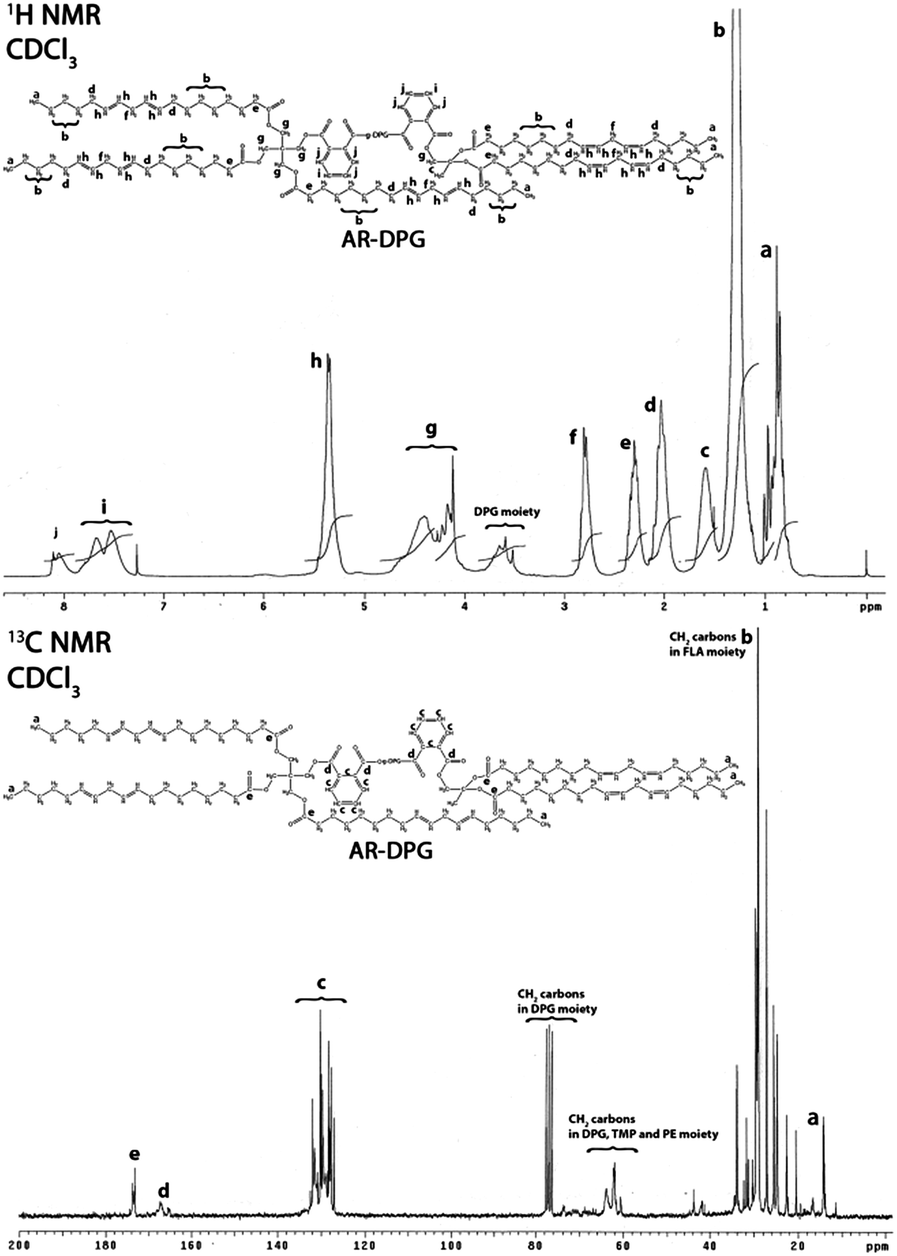 | ||
| Fig. 3 1H and 13C NMR spectra of AR-DPG (ith assignment of appropriate hydrogen and carbon in the most probable structure of synthesized AR-DPG). | ||
Results of 1H and 13C NMR analysis of AR-PE resin:
1H NMR: 0.88–1.11 (15H, m, 5 × C3, LFA moiety), 1.26 (70H, m, 35 × C2, LFA moiety), 1.60 (3H, m, 1 × C3, TMP moiety), 2.00–2.02 (20H, m, 10 × C2–CHCH–, LFA moiety), 2.26–2.32 (10H, m, 5 × C2–COO–), 3.58 (12H, m, 6 × C–C2–OH, PE moiety), 3.65 (4H, m, 4 × C–C2–OH, PE moiety); 4.15 (4H, m, 2 × COOC2), 4.17 (10H, m, 5 × C2–, TMP and PE moiety), 5.34 (20H, m, 10 × –CC–, LFA moiety), 7.26 (12H, m, HPh).
13C NMR: 14.12–18.00 (6 × H3), 22.56–33.99 (H2 carbons in FLA moiety), 48.0–49.2 (–(CH2–OH)3, PE moiety); 61.00–63.85 (H2 carbons in TMP and PE moiety), 127.20–128.50 (8 × CPh), 131.8 (2 × Ph()–COO, PA moiety), 132.9 (4 × Ph()–COO), 167.30 (O–Ph–O), 173.2 (O–Ph–O, PA moiety);
AR-G resin: 1H NMR: 0.88–0.94 (15H, m, 5 × C3, LFA moiety), 1.25 (70H, m, 35 × C2, LFA moiety), 1.51–159 (3H, m, 1 × C3, TMP moiety), 2.03–2.05 (20H, m, 10 × C2–CHCH–, LFA moiety), 2.26–2.34 (10H, m, 5 × C2–COO–), 2.77–3.01 (10H, m, 5 × –CHCH–C2–CHCH–, LFA moiety), 3.63–3.81 (10H, m, 2 × C2 and 2 × C, GLY moiety), 4.14–4.21 (4H, m, 2 × COOC2), 4.21–4.40 (10H, m, 5 × C2–, TMP and PE moiety), 5.34–5.36 (20H, m, 10 × –CC–, LFA moiety), 7.26 (2H, m, HPh, PA), 8.06 (7H, m, HPh).
13C NMR: 14.05 and 19.68 (6 × H3), 22.65–34.03 (H2 carbons in FLA moiety), 63.30–66.62, 76.37–77.65 (H2 carbons in GLY moiety), 63.30 (H2 carbons in TMP and PE moiety), 125.99–128.28 (8 × CPh), 131.54 (OC–HH–CO, PA moiety), 132.0 (4 × Ph()–COO), 167.26 (O–HCCH–O), 173.4 (O–HCCH–O, PA moiety).
Similar results were obtained for AR-PG, AR-DEG, AR-TME and AR-TMP.
The aesthetic appearances of synthesized AR were investigated (Fig. 4). The colors of the synthesized resins were slightly darker compared to the commercial ones, partly due to the brownish color of the PET glycolyzates used as reactants. Additionally, the darker color of the alkyds could be caused by several factors, such as the effect of residual catalysts (catalyst used for PET synthesis), transesterification catalyst (catalyze side reactions), residual of processing aids (catalytic or thermal transformation including incorporation into AR structure) and the severity of high temperature transesterification/esterification (condensation of by-product or products of side reactions – dehydration/condensation).30 Such complex reaction conditions contribute to the number of side reactions and secondary transformations which produce colored condensation products at different extent. The same amount of catalyst was used in all esterification reactions, thus its effect on the color development was similar for all obtained AR. Also, nitrogen was used as the inert gas throughout all reactions, indicating negligible influence of oxidation processes. The main reason for the darker color was due to the severity of tranesterification/esterification reactions together with successive dehydration/condensation of alcohols/glycolizate at high temperature. The polyesterification reactions, except of formation of linear/branched polymer, may result in a cyclic structure, which is mostly observed as chain terminating reaction. Probable explanation for lower average molar masses of AR-PE (Table 4) could be based on formation of higher extent of end cyclic structure, which terminated chain propagation. Polyol decomposition could be of appropriate significance which causes functionality decrease and thus increased contribution of termination reaction could also cause lower average molar mass. Dehydration of glycerin is well known reaction which produces acrolein, and subsequent addition reaction with another molecule of glycerine produces 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 allyliden glycerol.31 Exemplified side reaction causes reactant consumption and thus lower molar mass of resin and darker shade development as a consequence.
:2 allyliden glycerol.31 Exemplified side reaction causes reactant consumption and thus lower molar mass of resin and darker shade development as a consequence.
 | ||
| Fig. 4 The color of the alkyd resins synthesized from glycolyzate based on waste PET. | ||
| Alkyd code |
|
|
PDI |
|
The percent of difference between
|
|---|---|---|---|---|---|
| AR-DEG | 1558 | 7756 | 4.98 | 4747 | 205 |
| AR-PG | 2094 | 10163 |
4.85 | 4663 | 123 |
| AR-DPG | 1376 | 6578 | 4.78 | 4821 | 250 |
| AR-G | 2248 | 12342 |
5.49 | 4718 | 110 |
| AR-TME | 3507 | 13207 |
3.77 | 5446 | 55 |
| AR-TMP | 3187 | 11958 |
3.75 | 6079 | 91 |
| AR-PE | 1748 | 12458 |
7.13 | 4915 | 180 |
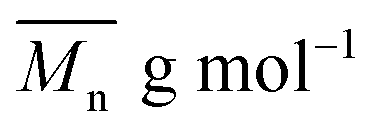
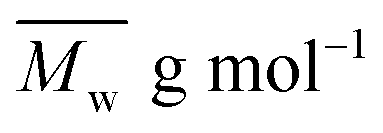
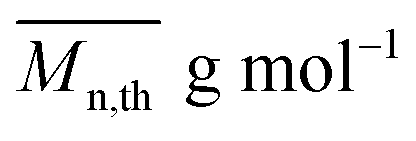
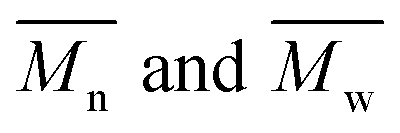
The average molar masses and molar mass distribution are major parameters that control polymer characteristics, both in the solid form and in solution.
In polymer science there has always been a desire for the better controlling of polymerization in order to produce polymers with the desired structure and narrower molar mass distribution. Because of the complexity of the reactants, alkyd resins normally have very broad molar mass distribution. Although, it is well-known that low molar mass components have a positive effect on adhesion, flexibility and gloss, while high molar mass components improve the drying rate, hardness, and solvent resistance,32 the best properties are achieved in alkyds with a narrow molar mass distribution.31
The average molar masses and molar mass distributions of the synthesized alkyd resins were determined by GPC. The obtained GPC molar mass distribution curves are presented in Fig. 5, while the calculated values of 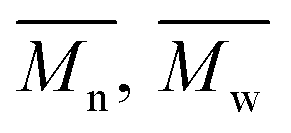 and the polydispersity index (PDI) are listed in Table 4.
and the polydispersity index (PDI) are listed in Table 4.
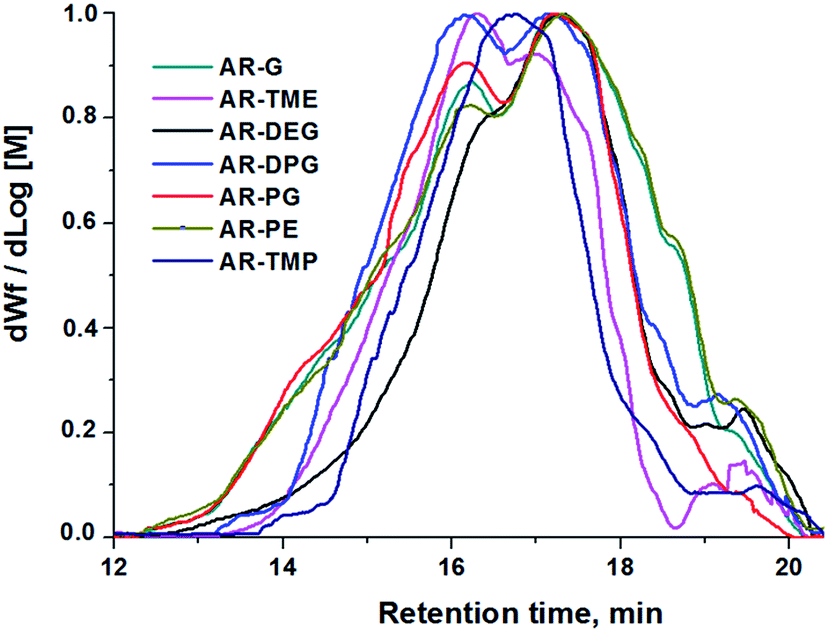 | ||
| Fig. 5 GPC chromatograms of the synthesized alkyd resins. | ||
Bearing in mind that the amounts of LFA, PA, PE and TMP in the reaction mixture were similar for all synthesized alkyd resins, the results of GPC analysis clearly evidence the effect of glycolyzate structure on the average molar masses and molar mass distributions of the alkyd resin samples. Alkyd resins prepared from difunctional glycolyzates (g-DEG, g-DPG and g-PG) had the lowest values of the average molar mass. Their polydispersity indices are in the range from 4.78 to 4.98 and are very close to the previously reported ones for conventional alkyd resins.33 Low values of the average molar masses for these resins were expected, as g-DPE, g-DPG and g-PG had only two hydroxyl groups. The low functionality of these glycolyzates lowered the overall branching and, consequently, the molar mass. These results further strengthened our confidence that difunctional glycolyzates should not be used as the alcohol component for the synthesis of alkyd resins. The average molar mass of alkyd resins prepared from tri-functional glycolyzates range from 2248 to 3507 g mol−1. Higher average molar masses of the resins prepared from tri-functional glycolyzates compared to the resins prepared from di-functional ones are due to the pronounced branching. Intriguingly, the polydispersity indices of the resins prepared from g-TME and g-TMP are lower compared to the alkyds prepared form g-G. These results could be explained by the fact that g-TME and g-TMP had only primary hydroxyl groups, while g-G had primary and secondary hydroxyl groups. The secondary hydroxyl groups of g-G were less reactive and, therefore, branching was less pronounced. Resins prepared from g-PE have unexpectedly low values of 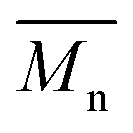 (1748 g mol−1) compared to the other resins synthesized from polyfunctional glycolyzates (2248, 3187 and 3504 g mol−1 for g-G, g-TMP and g-TME, respectively). The reasons for this rather contradictory result have not been yet entirely understood. A possible explanation for this could be attributed to the steric hindrance of hydroxyl groups at the glycolyzate chain-end, which retards the esterification reaction of all hydroxyl groups, thus effectively lowering the functionality of the PE glycolyzate. The high hydroxyl number of AR-PE supports this claim.
(1748 g mol−1) compared to the other resins synthesized from polyfunctional glycolyzates (2248, 3187 and 3504 g mol−1 for g-G, g-TMP and g-TME, respectively). The reasons for this rather contradictory result have not been yet entirely understood. A possible explanation for this could be attributed to the steric hindrance of hydroxyl groups at the glycolyzate chain-end, which retards the esterification reaction of all hydroxyl groups, thus effectively lowering the functionality of the PE glycolyzate. The high hydroxyl number of AR-PE supports this claim.
From the presented data it was clear that AR-TME and AR-TMP have the highest values of molar mass and the lowest values of PDI, anticipating the best applicative properties. The values of 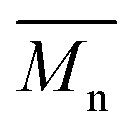 can be theoretically predicted by end-group analysis
can be theoretically predicted by end-group analysis 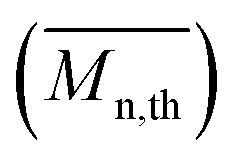 ,34,35 using the eqn (1):
,34,35 using the eqn (1):
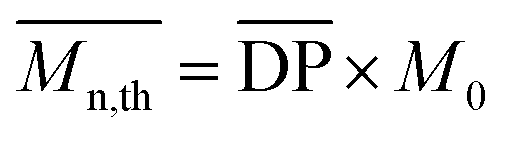 | (1) |
 is the mean degree of polymerization and M0 is the mean molar mass of the repeating unit of the alkyd resin.
is the mean degree of polymerization and M0 is the mean molar mass of the repeating unit of the alkyd resin.  could be calculated from the acid number of the synthesized alkyd resins by the eqn (2):
could be calculated from the acid number of the synthesized alkyd resins by the eqn (2):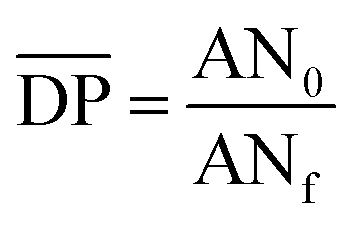 | (2) |
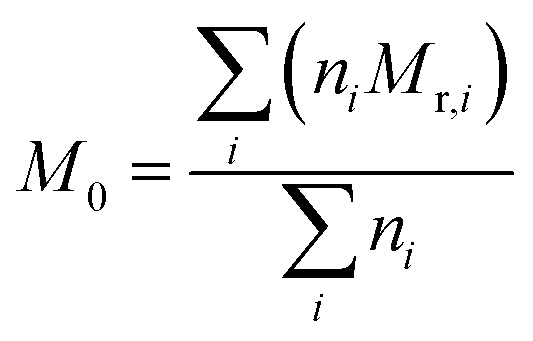 | (3) |
The calculated theoretical values of the average molar masses of the synthesized alkyd resins are presented in Table 4. The data from Table 4 showed a very large difference between 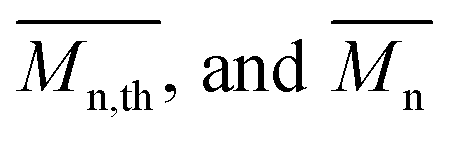 obtained by GPC ranging from 55% (g-TME) to 250% (g-DPG). Higher values for
obtained by GPC ranging from 55% (g-TME) to 250% (g-DPG). Higher values for 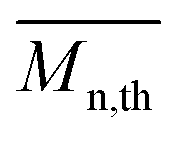 previously reported by many authors,36–38 were most likely due to the presumptions made in the end-group analysis. These presumptions did not take into account the influence of the polymer chain size on its reactivity, the intramolecular reaction and the different reactivity of the same functional groups.36,37 The low values of
previously reported by many authors,36–38 were most likely due to the presumptions made in the end-group analysis. These presumptions did not take into account the influence of the polymer chain size on its reactivity, the intramolecular reaction and the different reactivity of the same functional groups.36,37 The low values of 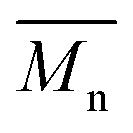 compared to
compared to 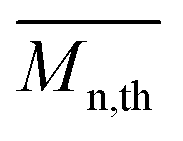 for AR-DPG and AR-DEG are probably a consequence of the structure of these glycolyzates. Namely, the long and flexible chain of these glycolyzates increased the chance of forming cyclic structures via intramolecular reactions. These reactions led to a decrease in the acid number and an increase in the consequently calculated
for AR-DPG and AR-DEG are probably a consequence of the structure of these glycolyzates. Namely, the long and flexible chain of these glycolyzates increased the chance of forming cyclic structures via intramolecular reactions. These reactions led to a decrease in the acid number and an increase in the consequently calculated  , while there was no corresponding increase in
, while there was no corresponding increase in 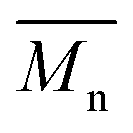 .
.
Understanding the solution behavior of resins is essential for adjusting the properties of the synthesized alkyds for optimal performances. Their practical usefulness can be realized only in solution in appropriate solvent systems. For instance, the solution viscosity of surface coatings determines their mode of application either by brushing or spraying and is equally important in dry film formation. After synthesis, all alkyd resins were diluted with xylene to 60% solid content. Resins AR-TME and AR-TMP, at this solid content, still had a too high viscosity, so these resins were further diluted to 50% solid content. The diluted resins reached a fluidity that could be cast into film easily. Thus, transparent and homogeneous films were obtained from all alkyd solutions in xylene. The values of the viscosity of all resins at 60% solid content are presented in Table 5. The viscosity tended to increase as the functionality of the glycolyzates increased. This could be explained by the fact that the resins synthesized from glycolyzates with higher functionality had a higher degree of branching and molar mass, and thereby higher viscosity. Even though AR-PE had lower molar mass compared to AR-G, it had a higher viscosity value. One possible explanation of this behavior could involve the tendency of the AR-PE macromolecules to form super-molecular aggregates, as a result of the large number of hydroxyl groups. These super-molecules could greatly increase the viscosity. Resin AR-DPG had a slightly lower viscosity compared to AR-DEG because the DPG glycolyzates had one more CH3 group in its molecule, which decreased the resistance to flowing.
| Alkyd code | Hardness | Dry-to-touch time, h | Viscosity, mPa s |
|---|---|---|---|
| AR-DEG | 39 | 10 | 4290 |
| AR-PG | 55 | 12 | 7020 |
| AR-DPG | 41 | 10 | 2160 |
| AR-G | 59 | 6 | 9270 |
| AR-TME | 86 | 6 | 56789 |
| AR-TMP | 78 | 6 | 43578 |
| AR-PE | 44 | 8 | 20300 |
As alkyd resins are exploited in film form, it was essential to investigate the effect of the functionality and the structure of the obtained glycolyzates on the key applicative properties of the product in this form. The drying mechanism of an alkyd coating is complex and is usually separated in two stages. The first stage involves the evaporation of the solvent and the second is the oxidative drying of the fatty acid chains, which eventually results in the formation of a polymer network. Many factors affect the drying rate and film formation, such as, the degree of unsaturation, the drying catalyst, the molar mass and molar mass distribution, the viscosity, the solvent, the amount of conjugated double bonds, the anti-skinning agent, etc. In our case, all of the aforementioned parameters were the same except the molar mass and viscosity. As we mentioned earlier, the higher molar mass component of the alkyd resin enhances the drying rate (physical), therefore, the alkyd resins prepared from the multifunctional glycolyzates showed better drying properties (Table 5). Although AR-PE had higher molar mass components (Fig. 5), it showed a longer drying time compared to AR-G, AR-TMP and AR-TME, probably due to the solvent retention. Namely, during the drying (solvent evaporation and resin cross-linking) of the very high molar mass fractions, solvent is entrapped in the coating due to the lower solvent diffusivity in highly viscous media.31
The dry-to-touch times, hardness and viscosity of the synthesized alkyd resins are presented in Table 5.
The hardness of the coatings was determined by the König pendulum method. The values of the hardness are given by the number of oscillations made by the pendulum within the specified limits of amplitude. This test is based on the principle that the amplitude of the pendulum's oscillation will decrease more quickly when supported on a softer surface. It is a well known fact that the molar mass of the resin, the amount of fatty acids (i.e. the cross-linking degree) and the reactants with ring structure mostly affect the hardness of the coating film. Bearing in mind that the amounts of PA and LFA were very similar in the reaction mixture (Table 1), it could be presumed that the molar mass and the molar mass distribution had the key influence on the coating hardness.
Fig. 6 shows the dependence of the coating hardness on the molar mass. The obtained linear correlation between coating hardness and the molar mass further confirmed our presumption that the molar mass had the major effect on the hardness of the coatings. Hardness values of AR-DEG, AR-PG and AR-DPG clearly indicate that glycolyzates based on DPG and DEG, i.e. g-DPG and g-DEG, contribute to lower film strength. Oppositely, incorporation of monomeric alcohol unit, i.e. PG in g-PG, contributes to significantly higher film hardness (27% increases) of AR-PG. Such results strongly indicate that inclusion of etheral linkage in alkyd resin significantly deteriorates film properties. On the other hand, the inclusion of AR-TMP and AR-TME in resin gave the highest hardness of obtained film, while oppositely, alkyd resin film based on g-G showed the lowest hardness among tetra-functional glycolyzate. The tetra-functionality of g-TME and g-TMP glycolyzates results from four primary hydroxyl groups, while in g-G two primary and two secondary low reactive groups are present. From the 1H NMR analysis, minor fraction (less than 8%) of g-G product with secondary hydroxyl group participation in ester group formation was found. Lower reactivity of two secondary group in g-G in a transesterification reactions during alkyd resin synthesis influences that g-G behave as di-functional g-DEG and g-DPG glycolyzates.
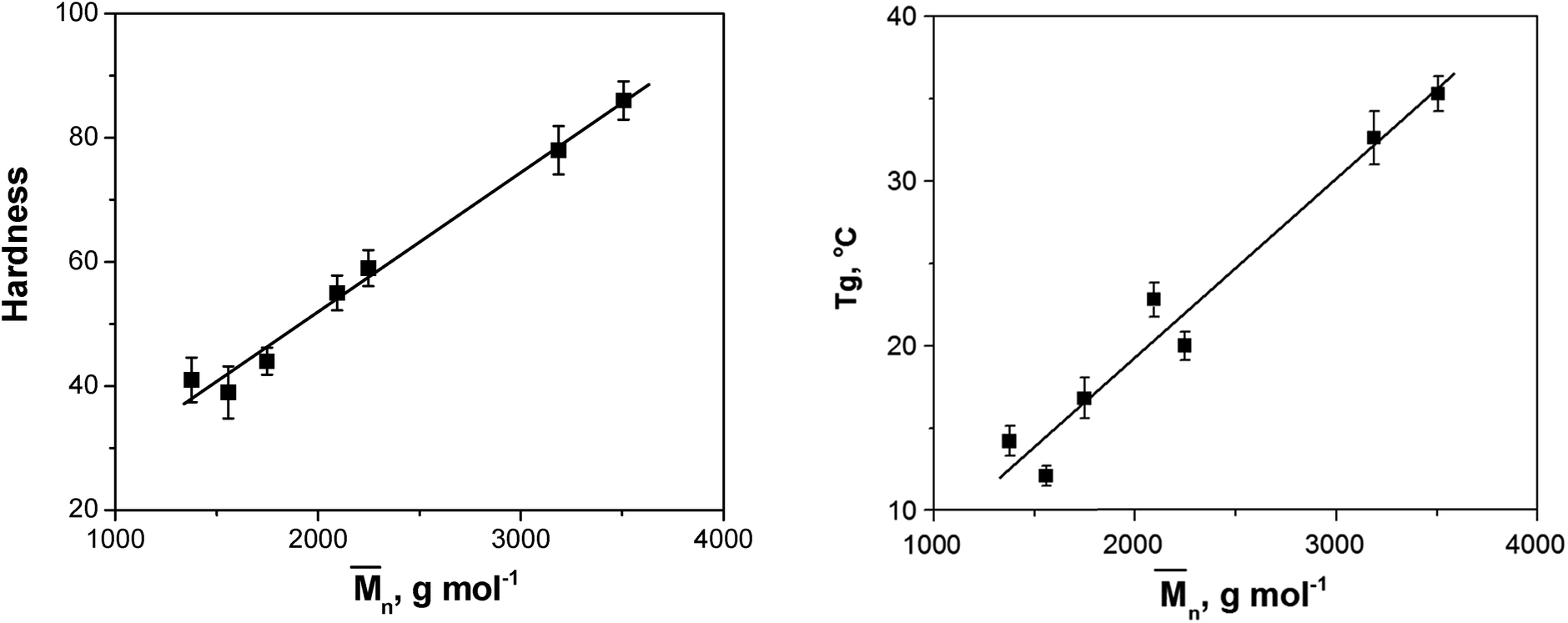 | ||
| Fig. 6 Dependence of hardness and glass transition temperature on the mean molar mass of the synthesized alkyd resins. | ||
The glass transition temperature is also a feature closely linked to hardness. The increase in Tg value is usually due to an increase in the molar mass, the cross-linking density, the quantity of short rigid groups (terephthaloyl moiety), the chemical structural order (symmetry), the polarity and the increasing hydrogen bond content. In this case, it could be assumed that the cross-linking density, the quantity of short rigid groups and the chemical structural order (symmetry) were similar in all synthesized alkyd resins. Although there were differences in the polarity and possible hydrogen bonding sites between the synthesized alkyd resins, it could be concluded by analyzing the obtained results for the Tg values that the molar mass had the highest effect on the Tg. For example, the alkyd resin prepared using PE glycolyzate had the highest hydroxyl group content (HV = 84 mg KOH per g), but its Tg value was lower compared to the Tg of AR-TMP and AR-TME (Table 6), which had lower HV values but higher molar masses.
| Alkyd code | Gloss, GU | Flexibility | Film thickness, μm | Adhesion | T g, °C |
|---|---|---|---|---|---|
| AR-DEG | 99.1 | 10 | 47.6 | 5 | 12.1 |
| AR-PG | 95.7 | 10 | 51.3 | 5 | 22.8 |
| AR-DPG | 96.3 | 10 | 54.4 | 5 | 14.2 |
| AR-G | 99.8 | 10 | 59.7 | 5 | 20.0 |
| AR-TME | 97.4 | 10 | 64.8 | 5 | 35.3 |
| AR-TMP | 97.3 | 10 | 55.7 | 5 | 32.6 |
| AR-PE | 94.2 | 10 | 49.7 | 5 | 16.8 |
The dependence of the Tg on the mean molar mass is presented in Fig. 6. The same as for hardness, the obtained correlation was linear.
From the aforementioned results it was clear that the use of TMP and TME glycolyzates was crucial for the synthesis of alkyd resins with higher mean molar mass and, therefore, improved hardness.
All coatings showed no cracking or delamination when subjected to the cupping tester (Table 6). This result indicated that the cross-linked films were relatively flexible and adherent at the coating thickness utilized. Often, coatings with excellent hardness fail the flexibility test, so it is important to emphasize that even the samples with very high hardness values (rigid film) passed the flexibility test.
The gloss property is important when the esthetic or decorative appearance of a coating is of major significance. As presented in Table 6, the glosses measured at 60° were found to be excellent. Furthermore, since the gloss is influenced by the roughness and the texture of the film surface, the excellent gloss obtained for all synthesized resins, confirmed homogeneous drying/cross-linking throughout the thickness of the film and good compatibility of the reactants (PET glycolyzates, PE, TMP, FAL and PA), which caused good film-forming and leveling properties.
The adhesion of the films to the substrate was characterized using the crosshatch adhesion method. With this method, a rating system was used to convey the degree of adhesion loss.
A rating of 0 indicated the removal of more than 65% of the coating from the test area, while a rating of 5 indicated no coating removal. The reproducible adhesion test results showed that all synthesized alkyd resins had an excellent adhesion to untreated metal plates (Table 6).
These results could be explained by the fact that the synthesized resins had a high number of terminal hydroxyl groups, which could interact with the metal surface.
Excellent adhesion indicated homogeneous drying/cross-linking by auto-oxidation throughout the thickness of the film. If drying of the film is inhomogeneous, internal stress often occurs due to the fact that a higher oxygen concentration at the coating/air interface leads to faster drying of the coating surface. Coatings with moderately high internal stress are more susceptible to adhesion failure because the molecular rearrangements required to reduce the stress can also disrupt adhesive interactions at the coating/substrate interface.39
As shown in Table 7, all examined coatings were totally resistant to distilled water and 2% NaCl solution. Generally, all synthesized alkyds were unaffected by the acidic solution, except for a slight color change. The water and acid resistance of a coating are very important to ensure high coating durability during use. When applying the NaOH resistance test, it was observed that an alkaline environment strongly affected the coated films of all alkyds, except AR-TMP and AR-TME, causing some swelling, color change and film blistering. The poor alkaline resistance of these alkyds was attributed to the presence of alkali-hydrolysable ester linkages in the alkyd backbones of the resin. AR-TMP and AR-TME were far less affected by alkali solution. The high alkaline and acidic resistance of the AR-TMP and AR-TME coatings could be explained by the high cross-link density of their networks, which decreased the exposure to the environment of the inner parts of the film. Xylene was used to evaluate the non-polar solvent resistance. Coatings prepared from multifunctional glycolyzates showed excellent xylene resistance, while coatings prepared from difunctional glycolyzates faintly swelled. The excellent xylene resistance of the coatings indicated that the solvent was not trapped in the dried film. Polar solvents, such as acetone, are regularly used to estimate the degree of the cure of the prepared resin. If the film is less affected, that means that the solvent molecules find it harder to penetrate the cross-linked network, indicating less free volume and that segmental mobility was retained in the polymer. The latter means a higher degree of overall cross-linking of the resin and better properties.
| Alkyd code | Chemical medium | |||||
|---|---|---|---|---|---|---|
| Distilled water | NaCl (2 wt%) | NaOH (2 wt%) | H2SO4 (2 wt%) | Xylene | Acetone | |
| a 1 – completely unaffected, 2 – unaffected, slightly color changed, 3 – film faintly swelled, 4 – film slightly cracked and blistered, 5 – film cracked and removed. | ||||||
| AR-DEG | 1 | 1 | 5 | 2 | 3 | 5 |
| AR-PG | 1 | 1 | 4 | 2 | 3 | 5 |
| AR-DPG | 1 | 1 | 4 | 2 | 3 | 5 |
| AR-G | 1 | 1 | 4 | 2 | 1 | 5 |
| AR-TME | 1 | 1 | 2 | 1 | 1 | 4 |
| AR-TMP | 1 | 1 | 2 | 1 | 1 | 4 |
| AR-PE | 1 | 1 | 4 | 2 | 1 | 5 |
Conclusion
High-performance alkyd resins were prepared using the multifunctional glycolysis products of postconsumer PET bottles. The properties of the alkyd resins were found to be highly dependent on the functionality and the structure of the employed glycolyzates. Generally, alkyd resins with better overall properties were derived from the multifunctional glycolyzates. Considering all the conducted investigations, the tetrafunctional TME and TMP glycolyzates gave alkyd resins with the best properties. Both glycolyzates had four sterically free (unshielded), primary OH groups capable of interacting and forming alkyds with a higher degree of overall branching and, therefore, higher molar mass of AR-TME and AR-TMP were obtained. The latter provided improved mechanical, drying and chemical resistance properties compared to the reported alkyds from difunctional glycolyzates and also conventional general purpose resins. Tetra-functional glycolyzate g-G, the same functionality as g-TME and g-TMP, and hexa-functional g-PE glycolyzate, led to the poorer applicative properties of the corresponding resin due to the specificity of their structures. In the first case, two secondary, less reactive OH groups, and in the second case, the steric hindrance of the hydroxyl groups at the glycolyzate chain-end decreased the branching degree.Future work on this matter will involve the preparation, analysis and application of lacquers, paints and anticorrosive coatings based on the presented alkyd resins.
Acknowledgements
The authors would like to acknowledge funding from the Ministry of Education, Science and Technological Development of the Republic of Serbia, through Project No. 172062 “Synthesis and characterization of novel functional polymers and polymeric nanocomposites”.References
- M. Soucek and M. K. G. Johansson, Prog. Org. Coat., 2012, 73, 273 CrossRef CAS.
- A. Hofland, Prog. Org. Coat., 2012, 73, 274–282 CrossRef CAS.
- F. Welle, Resour., Conserv. Recycl., 2011, 55, 865–875 CrossRef.
- https://www.smitherspira.com/market-reports/news/packaging/demand-for-pet-packaging-material-by-2019.aspx, (accessed December 2014).
- http://www.packagingnews.co.uk/news/global-consumption-of-pet-packaging-forecast-to-grow-to-over-20m-tonnes-by-2019/, (accessed December 2014).
- S. R. Shukla, A. M. Harad and L. S. Jawale, Polym. Degrad. Stab., 2009, 94, 604–609 CrossRef CAS.
- M. Kathalewar, N. Dhopatkar, B. Pacharane, A. Sabnis, P. Raut and V. Bhave, Prog. Org. Coat., 2013, 76, 147–156 CrossRef CAS.
- M. N. Siddiqui, H. H. Redhwi and D. S. Achilias, J. Anal. Appl. Pyrolysis, 2012, 98, 214–220 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- L. Bartolome, M. Imran, K. G. Lee, A. Sangalang, J. K. Ahn and D. H. Kim, Green Chem., 2014, 16, 279–286 RSC.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- F. Pardal and G. Tersac, Polym. Degrad. Stab., 2007, 92, 611–616 CrossRef CAS.
- S. M. Cakić, I. S. Ristić, M. M. Cincović, N. Č. Nikolić, O. Z. Ilić, D. T. Stojiljković and J. K. B. Simendić, Prog. Org. Coat., 2012, 74, 115–124 CrossRef.
- A. Torlakoğlu and G. Güçlü, Waste Management, 2009, 29, 350–354 CrossRef PubMed.
- A. Marinković, T. Radoman, E. Džunuzović, J. Džunuzović, P. Spasojević, B. Isailović and B. Bugarski, Hem. Ind., 2013, 67, 913–922 CrossRef.
- L. Bartolome, M. Imran, B. G. Cho, A. A. M. Waheed and H. K. Do, Recent developments in the chemical recycling of PET, in Material Recycling – Trends and Perspectives, ed. D. S. Achilias, Intech, Croatia, 2012, pp. 65–84 Search PubMed.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312–320 CrossRef.
- G. Güçlü and M. Orbay, Prog. Org. Coat., 2009, 65, 362–365 CrossRef.
- G. Güçlü, Polym. Bull., 2010, 64, 739–748 CrossRef.
- Ö. Tuna, A. Bal and G. Güçlü, Polym. Eng. Sci., 2013, 53, 176–182 Search PubMed.
- J. Dullius, C. Ruecker, V. Oliveira, R. Ligabue and S. Einloft, Prog. Org. Coat., 2006, 57, 123–127 CrossRef CAS.
- ASTM D1957-86R01, Test Method for Hydroxyl Value of Fatty Oils and Acids.
- T. C. Patton, Alkyd resin technology: formulating techniques and allied calculations, Interscience, New York, 1962, pp. 93 Search PubMed.
- D. B. Pourreau and S. E. Smyth, 81st Annual Meeting of the Federation of Societies for Coatings Technology, Philadelphia, 2004 Search PubMed.
- R. Xiong, S. I. Sandler, D. G. Vlachos and P. J. Dauenhauer, Green Chem., 2014, 16, 4086–4091 RSC.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- ASTM D1639, Test Method for Acid Value of Organic Coating Materials.
- T. Mizutani, K. Arai, M. Miyamoto and Y. Kimura, Prog. Org. Coat., 2006, 55, 276 CrossRef CAS.
- V. Atimuttigul, S. Damrongsakkul and W. Tanthapanichakoon, Korean J. Chem. Eng., 2006, 23, 672–677 CrossRef CAS.
- K. Holmberg, High Solids Alkyd Resins, Marcel Dekker, INC, New York, 1st edn, 1987, ch. 2, pp. 39 Search PubMed.
- H. J. Konig, Farbe Lack, 1978, 84, 756 Search PubMed.
- K. Manczyk and P. Szewczyk, Prog. Org. Coat., 2002, 44, 99 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3096–3099 CrossRef CAS.
- L. C. Case, J. Polym. Sci., 1957, 26, 333–350 CrossRef CAS.
- M. Jonaso, J. Appl. Polym. Sci., 1960, 4, 129–140 CrossRef.
- X. Yin, H. Duan, X. Wang, L. Sun, W. Sun, H. Qi and L. Ma, Prog. Org. Coat., 2014, 77, 674–678 CrossRef CAS.
- L. W. Hill, J. Coat. Technol., 1992, 64, 29–41 CAS.
| This journal is © The Royal Society of Chemistry 2015 |