
Enthalpy–entropy relations in the acid–base equilibrium of warfarin and 10-hydroxywarfarin; joint experimental and theoretical studies†

Abstract
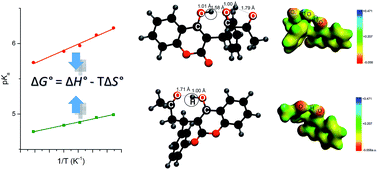
In this work we delineate basic thermodynamic factors that govern the acid–base equilibrium of the phenolic drug warfarin, pKa = 4.99, and its metabolite 10-hydroxywarfarin, pKa = 5.95. By applying experimental and theoretical approaches we have determined the enthalpic and entropic contributions to the dissociation free energy for both molecules. We have found that formation of specific intramolecular hydrogen bonds: OH⋯O by warfarin and OH⋯OH⋯O by 10-hydroxywarfarin, respectively, may be of a great importance for the changes in enthalpy and entropy during dissociation. The Car–Parrinello molecular dynamics have shown that these bonds are present at both temperatures T = 298 K and T = 378 K. We infer that an uncommon double bridge of 10-hydroxywarfarin, OH⋯OH⋯O, imposes lower flexibility on the molecule and thereby a stronger rise of heat capacity and enthalpy upon dissociation than the single OH⋯O bond noted for warfarin. It has been proposed, in addition, that the entropic contributions derive from the two opposite effects related to: solvation of ions – it leads to a loss of entropy, and breaking of intramolecular bonds – it causes a gain of entropy. For 10-hydroxywarfarin these effects seem to be equal in magnitude, and thereby, its dissociation entropy is close to zero. Furthermore, we show that addition of a surfactant and aprotic cosolvent changes the enthalpy–entropy relations and induces the upward pKa shifts of both molecules, up to 1.5 pH unit. The changes in thermodynamics which are caused by these two additives are, however, totally different.
 Please wait while we load your content...
Please wait while we load your content...

