
A greener approach towards double heteroarylation of N, O and S nucleophiles: synthesis of bioactive polynuclear fused N-heteroarenes†
Abstract
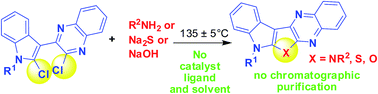
A catalyst, ligand and solvent free method for double heteroarylation of N, O and S nucleophiles has been developed for the first time leading towards the synthesis of compounds containing an indole ring fused with pyrrolo-, furo- and thieno[2,3-b]quinoxaline moieties. This general and greener approach afforded novel compounds of medicinal importance.
 Please wait while we load your content...
Please wait while we load your content...

