
Synthesis and properties of a millable polyurethane elastomer with low halloysite nanotube content
Bing Gong,
Chunfa Ouyang*,
Ye Yuan and
Qun Gao
School of Materials Science and Engineering, Shanghai Institute of Technology, 100 Haiquan Road, Shanghai 201418, China. E-mail: ouyoung_0916@163.com
First published on 7th September 2015
Abstract
Nanocomposites comprised of millable polyurethane (MPU) and very low loadings of halloysite nanotubes (HNTs) have been prepared with high tensile strength and elongation at break, and cured using either toluene diisocyanate dimer or dicumyl peroxide as crosslinking agents to further enhance the mechanical performance of the nanocomposites and to reduce costs. A 1.17-fold increase in tensile strength and a 0.58-fold increase in elongation at break was achieved for synthetic linear polyurethane elastomer nanocomposites by incorporating only 1 wt% of HNTs, and these increases were primarily attributed to the formation of covalent bonds between the OH groups of HNTs and the NCO groups of 4,4′-diphenyl-methane diisocyanate (MDI). Remarkable improvements in Young's modulus were further obtained after curing using the crosslinking agents. The nanostructure of these nanocomposites consisted of individual HNTs covalently bonded and specifically associated with hard MPU microdomains, and therefore affected the degree of phase separation, as characterized by spectroscopy studies and direct microstructural analysis. Moreover, an unprecedented environmental solvent-free prepolymer method has been proposed to compound the MPU/HNTs nanocomposites.
Introduction
Polyurethane (PU) constitutes one of the most important classes of polymeric materials, and is well-known for its excellent qualities. Because of its diverse utility and relative low cost, these materials account for nearly 5 wt% of the total worldwide polymer production, and their production is expected to exceed 18 kilotons annually by 2016.1,2 The basic constituents of PU elastomers are a diisocyanate, which is a long chain oligomeric polyol that may be a polyether or polyester oligomer, and low molecular weight diols or diamines as a chain extender.3 PU elastomers can be divided into three different types depending upon their structure, i.e., castable polyurethane (CPU), thermoplastic polyurethane (TPU), and millable polyurethane (MPU). MPU is a special type of synthetic rubber that is generally compounded by means of conventional rubber processing equipment in the presence of other ingredients and additives.4–7 MPU is of high demand in various applications such as footwear, as hoses, and oil field products owing to its high toughness, flexibility, strength, abrasion resistance, and chemical resistance. Many large industrial rolls, copier rolls, O-rings, seals, gaskets, and many other mechanical items requiring abrasion resistance are fabricated using these special elastomers.8–18Nanocomposites are typically defined as composites having more than a single solid phase with dimensions less than 100 nm.19–22 Over the past two decades, investigation and development of polymer nanocomposites have attracted great attention owing to the unique characteristics of nanoparticles such as their large surface area, high surface reactivity, and relative low cost.23–25 Previously, nanometer-sized materials with extremely large surface areas or a high aspect ratios, including graphite oxide nanoplatelets,26 polyhedral oligomeric silsesquioxane,27,28 carbon nanotubes,29–32 and layered silicate clays33–37 have been incorporated into CPU or TPU to enhance their mechanical performances and thermal stability. In the present study, halloysite nanotubes (HNTs) were chosen as the nanofiller, and are incorporated into MPU. Halloysite is a typical type of clay nanofiller naturally deposited in various countries around the world, such as the United States, China, New Zealand, France, and Belgium, that has recently attracted a great amount of attention. The material is defined as a two layered aluminosilicate, chemically similar to kaolin, but which has a predominantly hollow tubular structure in the submicron range.38 The external surface of the nanotube has a tetrahedral sheet structure that mainly consists of siloxane groups (Si–O–Si), while the internal surface has an gibbsite octahedral structure with aluminol groups (Al–OH).39 HNTs exhibit unique surface chemical properties because of the relatively small number of hydroxyl groups located on the external surface owing to the multi-layer structure.40 Therefore, relative to other layered silicates, halloysite can be more easily dispersed in a polymer matrix due to the weak secondary interactions among the nanotubes via hydrogen bonds and van der Waals forces. Moreover, the cost of HNTs is relatively low.41,42 In addition, HNTs have been incorporated into polymer matrices as environmentally benign reinforcement fillers because of their outstanding intrinsic properties such as nanoscale dimensions, high surface area, unique morphology, low density, high specific strength and Young's modulus, and very low coefficient of thermal expansion.43–47
Although a number of reports can be found in the literature regarding PU nanocomposites, no comprehensive study has thus far been undertaken for MPU nanocomposites. To the authors' knowledge, this is the first study to introduce very low loadings of HNTs into the synthesis of MPU to stiffen and strengthen the material, and various vulcanization processes have been conducted to further enhance the synthetic nanocomposite performance and reduce costs. In addition, an unprecedented environmental solvent-free prepolymer method has been proposed to compound MPU/HNTs nanocomposites.
Experimental section
Materials
Because the preparations involved toxic and reactive isocyanates, all the necessary precautions for their safe handling and for conducting their reactions under anhydrous conditions were taken in all the experimental work.4,4′-Diphenyl-methane diisocyanate (MDI, TCI), 1,4-butanediol (1,4-BD, Aldrich), 2,4-tolylene diisocyanate (TDI, YMIOO chemical), dicumyl peroxide (DCP, Aldrich), pyridine (99%, Sinopharm), calcium oxide (CaO, Aldrich), dioctyl phthalate (DOP, Aldrich), 1,3,5-tri-2-propenyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TAIC), and phosphoric acid (98%, Sinopharm) were used as received. Poly(propylene glycol) (PPG, Mn = 1000) and precipitated silica were respectively purchased from Shanghai JiaHua and NanJi Chemical Co., Ltd in China, and were used as received. HNTs with 15–20 nm inner lumen diameters, 30–50 nm external diameters, and approximate lengths of 100–1500 nm were purchased from Lingshou Longchuan drilling plugging materials factory (China) without any chemical modification.
Synthesis of crosslinking agent (TDID)
The dimer of TDI (TDID) was synthesized by adding 1 ml of freshly distilled pyridine to 50 ml of TDI in a round-bottomed flask under an N2 atmosphere. The reaction mixture was left undisturbed for 7 d. The precipitated crystals were filtered and washed several times with dry toluene to obtain white crystals of TDID.Synthesis of millable polyurethane elastomer nanocomposites
The reaction was conducted in a four-necked, 500 ml round bottom flask fitted with an agitator, a temperature indicator, and nitrogen inlet-outlet tubes. The PPG was dried under vacuum at 120 °C for 2 h prior to use. The polyether PU prepolymer was synthesized by reacting MDI (122 g) with PPG (165.7 g) and a drop of phosphoric acid (0.033 ml) in the presence of HNTs at 82 °C for 3 h. Subsequently, the chain extender 1,4-BD (31.35 g) was dropwise added to the prepolymer and stirred at 50 °C prior to casting and curing in a Teflon mold at 120 °C for 6 h to obtain the transparent film. The concentration of reactive hydroxyl groups on HNTs was 29 ± 3 mmol g−1, and was measured using titration to determine the excessive isocyanate groups after mixing HNTs with a known amount of MDI. The molar ratio of MDI to PPG in the prepolymer was 2.85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the total NCO/OH ratio in the PU was 0.95. Transparent PU nanocomposite films containing 0.5, 1, and 5 wt% HNTs were successfully prepared by varying the amount of HNTs and 1,4-BD, and are coded herein as MPU/HNT-0.5, MPU/HNT-1, and MPU/HNT-5, respectively. Pure PU film was prepared in the absence of HNTs, and is coded simply as MPU. The reaction procedure is illustrated as follows in Scheme 1. It should be noted that what is expressed as the hard segment in Scheme 1 was composed of MDI and 1,4-BD, whereas that denoted as the soft segment was PPG.
:1, and the total NCO/OH ratio in the PU was 0.95. Transparent PU nanocomposite films containing 0.5, 1, and 5 wt% HNTs were successfully prepared by varying the amount of HNTs and 1,4-BD, and are coded herein as MPU/HNT-0.5, MPU/HNT-1, and MPU/HNT-5, respectively. Pure PU film was prepared in the absence of HNTs, and is coded simply as MPU. The reaction procedure is illustrated as follows in Scheme 1. It should be noted that what is expressed as the hard segment in Scheme 1 was composed of MDI and 1,4-BD, whereas that denoted as the soft segment was PPG.
 | ||
| Scheme 1 Reaction scheme for the synthesis of elastomeric gum polyurethane nanocomposites. | ||
Processing of millable polyurethane elastomer nanocomposites
PU was masticated in an open twin roller mill. During this mastication process, the temperature was maintained below 60 °C by circulating cold water through the rollers. The samples were subjected to compression molding at optimum temperature and time that were determined from the rheometric results. The formulations of the two different curing systems employed are listed in Table 1 and 2. The vulcanized products obtained using DCP and TDID as the curing agents are coded herein as D-MPU/HNTs and T-MPU/HNTs, respectively.| Raw rubber (phr) | DCP (phr) | TAIC (phr) | CaO (phr) | Precipitated silica (phr) | DOP (phr) | |
|---|---|---|---|---|---|---|
| Sample | 100 | 3 | 2 | 3 | 30 | 3 |
| Raw rubber (phr) | TDID (phr) | CaO (phr) | Precipitated silica (phr) | DOP (phr) | |
|---|---|---|---|---|---|
| Sample | 100 | 8 | 3 | 30 | 3 |
Characterization
Fourier transform infrared (FTIR) spectra were obtained on an iZ™ 10 spectrometer (Nicolet, USA), and each spectrum was comprised of 16 scans with a resolution of 4 cm−1. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on an AVANCE III NMR spectrometer (Bruker Corp., Switzerland). Dimethyl sulphoxide (DMSO-d6) was used as the solvent, and the measurements were conducted at 600 MHz. The chemical shifts in the discussion are reported in ppm. X-ray diffraction (XRD) measurements were conducted on a D/max 2200 PC X-ray diffractometer (Rigaku Corp., Japan) equipped with a CuK radiation source (50.1540 nm, operated at 40 kV and 30 mA) over a 2θ range of 5° to 40° with a scan speed of 2° min−1. Thermogravimetric analysis (TGA) was conducted with an STA 499 F3 Jupiter® (NETZSCH, Germany) with N2 as the purge gas at a flow rate of 40 ml min−1. Samples (8–12 mg) were heated from room temperature to 700 °C at a heating rate of 10 °C min−1. Atomic force microscopy (AFM) images were obtained by an AFM instrument (Beijing Nano-Instruments, Ltd, China) operating in the tapping mode using silicon cantilevers (spring constant: 3–40 N m−1; cantilever resonant frequency: 75–300 kHz).Dynamic mechanical analysis (DMA) was conducted on a DMA 242 C dynamic mechanical analyzer (NETZSCH, Germany) over a temperature range of −100 °C to 170 °C at a heating rate of 3 °C min−1 and a frequency of 1 Hz. Transmission electron microscopy (TEM) was conducted using a JEM-2010 high resolution field emission microscope (Jeol, Ltd, Japan) operating at 200 kV, providing a lattice resolution of 0.14 nm. The mechanical properties of the products were measured following the GB/T 528-1998 standard using a SUN 500 universal testing machine (GALDABINI, Italy). The crosshead speed was 50 mm min−1. All measurements were repeated five times, and the averages were obtained. The hardness of the products were measured by a Shore A hardness tester following the ASTM D 2240-2004 standard. All hardness measurements in this report were conducted after 10 s when the indenter had penetrated into the composites.
Results and discussion
The FTIR spectra of HNTs, pristine MPU, various MPU/HNTs nanocomposites, and a control sample comprised of a mixture of HNTs and MDI, are shown in Fig. 1. To help investigate the interfacial interaction between HNTs and the MPU matrix, a control sample comprised of a mixture of HNTs and MDI was prepared by reacting MDI (10 g) with HNTs (2.759 g), and dispersing in DMAC (20 g) at 82 °C for 3 h. Subsequently, the solvent was evaporated at 100 °C for 6 h in a vacuum oven. The FTIR spectra of the pristine MPU exhibited characteristic bands of urethane stretching (N–H) at 3280 cm−1, a combination of urethane carbonyl (NH–CO–O) and esteric carbonyl (CO–O) at 1727 cm−1, and a combination of N–H out-of-plane bending and C–N stretching at 1550–1520 cm−1. Analogously, a combination of urethane carbonyl (NH–CO–O) and esteric carbonyl (CO–O) was detected at 1727 cm−1, and N–H out-of-plane bending was detected at 1516 cm−1 in the control sample as a result of the generation of urethane bonds, which confirmed the reaction between NCO groups of MDI and OH groups of HNTs, as illustrated in Scheme 2. In the HNTs, the O–H bonds of the inner hydroxyl groups and outer surface hydroxyl groups were detected at 3619 and 3695 cm−1, respectively. The HNTs infrared spectrum exhibited absorption bands around 1034 and 910 cm−1, which were attributed to the Si–O stretching vibrations and Al–OH vibrations, respectively.44 For all nanocomposite spectra, the intensities of the absorptions near 3695 and 3619 cm−1 greatly decreased, which was due to the reaction of the OH groups of HNTs with the NCO groups of MDI. Similarly, the NCO group band was not detected at 2300–2200 cm−1 as a result of complete reaction. The carbonyl absorption band split into two peaks at 1704 and 1727 cm−1, corresponding to hydrogen-bonded and free carbonyl groups, respectively. Aihua et al. reported an equivalent finding.48 The HNTs were covalently bonded to the PU molecular chains during the synthesis of the prepolymer, which affected the phase separation of the segmented polyether PU. | ||
| Fig. 1 FTIR spectra of HNTs, pristine MPU, MPU/HNTs nanocomposites (raw rubber), and a control mixture of HNTs and MDI. | ||
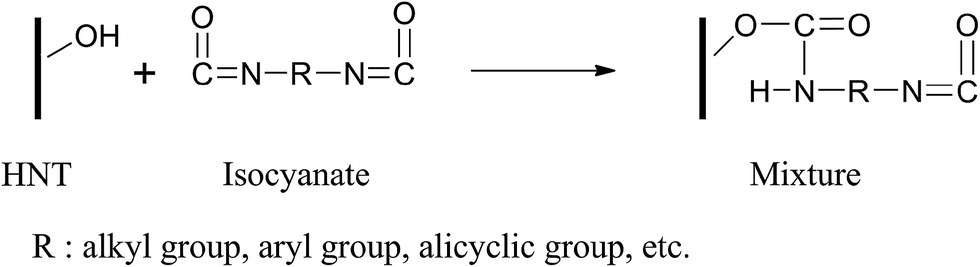 | ||
| Scheme 2 Reaction scheme between a single OH of an HNT and isocyanate. | ||
1H NMR spectra further confirmed the structure of the synthesized pristine MPU, as shown in Fig. 2(A). The urethane NH proton peak was observed at 9.5 ppm. The aromatic protons were observed between 7.1 and 7.4 ppm, and the aliphatic CH2 protons were observed at 1.2–4.2 ppm due to different chemical environments. The structure of the TDID was confirmed by 1H NMR spectroscopy, as shown in Fig. 2(B). The CH3 proton peak of TDI was observed at 2.01 ppm. The signals between 6.40 and 7.00 ppm were ascribed to the proton in the aromatic ring.
 | ||
| Fig. 2 1H NMR spectra of (A) pristine MPU and (B) TDID. | ||
The thermal stabilities of pristine MPU and MPU/HNTs nanocomposites was studied by TGA. As shown in Fig. 3, the TGA traces primarily indicate a two-step degradation pattern, which is consistent with the two peaks of the derivative thermogravimetry (DTG) curves shown in the inset of Fig. 3. The main results containing the onset temperature (Tonset) and the temperatures of 10% (T10%) and 50% (T50%) weight loss are presented in Table 3. The degradation of both MPU and MPU/HNTs nanocomposites occurred above 300 °C, and exhibited three ranges of weight loss: (i) decomposition due to water evaporation; (ii) decomposition of urethane linkages into isocyanate and alcohol with possible formation of primary and secondary amines; (iii) breaking of the ester bonds of the polyol soft phase of the polyurethane.49–52 No significant influence on the thermal stability of the MPU/HNTs nanocomposites can be attributed to the presence of HNTs.
 | ||
| Fig. 3 TGA thermograms of pristine MPU and MPU/HNTs nanocomposites. | ||
| Samples | Tonset (°C) | T10% (°C) | T50% (°C) |
|---|---|---|---|
| MPU | 312 | 324 | 360 |
| MPU/HNTs-0.5 | 303 | 316 | 355 |
| MPU/HNTs-1 | 306 | 319 | 363 |
| MPU/HNTs-5 | 300 | 315 | 360 |
Fig. 4 presents the tapping mode AFM phase images of the pristine MPU and MPU/HNTs nanocomposites. Here, the lighter regions correspond to the hard phase whereas the darker regions are representative of the polyol, i.e., the soft segments.53 On the surface of MPU, the soft segments exist in the concave regions because the soft molecular chains and easy motion form a continuous phase distribution that surrounds the hard segments, which exist in the convex regions because of the higher surface energy that forms the dispersed phase distribution. As HNTs were progressively added into the MPU, the hard segment regions became increasingly bright. These results indicate that the HNTs preferentially reinforced the hard microdomains rather than the soft segments of the MPU. Although the concentration of surface OH groups was small (due to the hollow tubular shape, the OH groups were mainly concentrated on the interior of the HNT structure and were also present on the edges of the nanotubes), particularly when compared to other aluminosilicates such as montmorillonite,54 the high polarity of the MPU matrix favored this interaction, and the formed interface was effective.
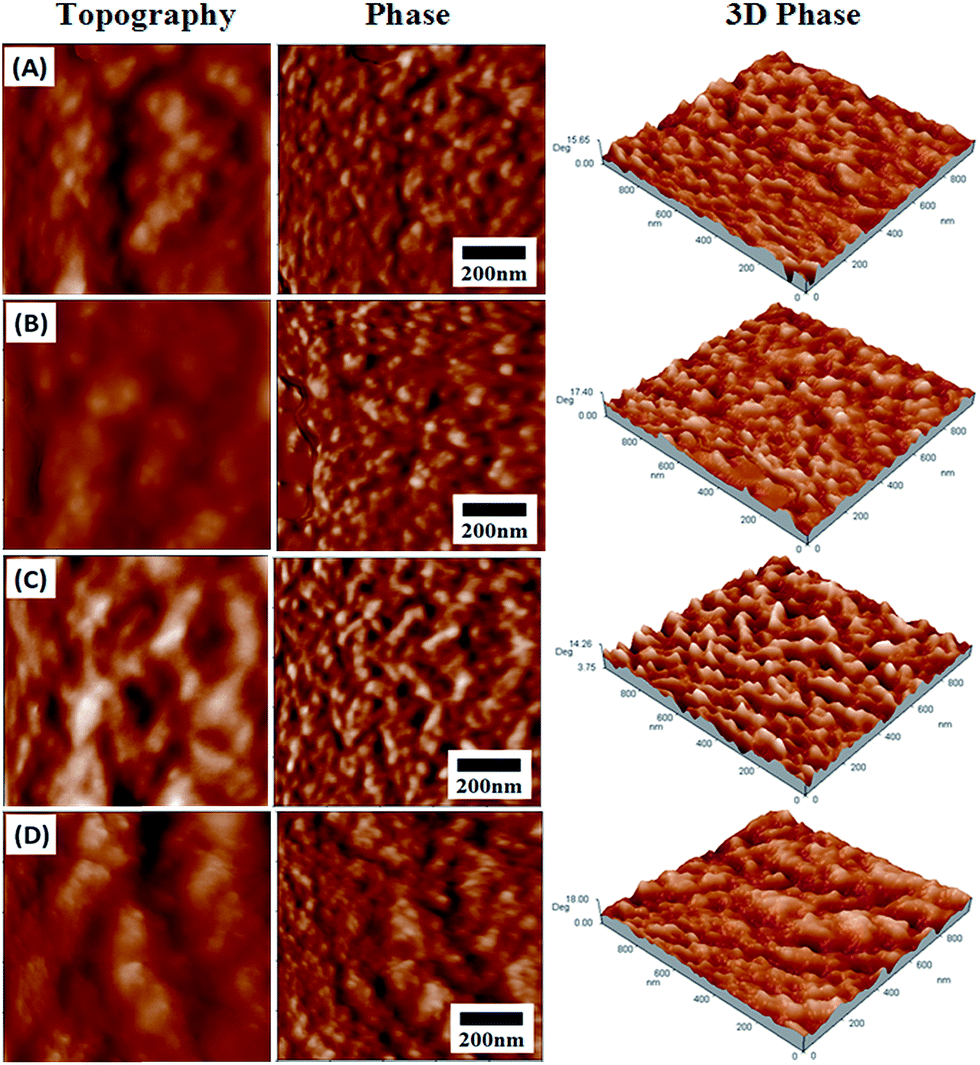 | ||
| Fig. 4 Surface topography (left), phase (middle), and three-dimensional phase (right) diagrams of pristine MPU and MPU/HNTs nanocomposites obtained by AFM: (A) MPU; (B) MPU/HNTs-0.5; (C) MPU/HNTs-1; and (D) MPU/HNTs-5. | ||
XRD is an important tool to characterize the structure of materials. The XRD spectra of HNTs, MPU, and the MPU/HNTs nanocomposites are presented in Fig. 5. As indicated in the figure, the pristine MPU matrix presents a very strong, broad reflection at about 2θ = 20.09°, corresponding to an amorphous material. For the MPU/HNTs nanocomposites, the new characteristic reflections at 2θ values of 12° and 30°, which correspond to the HNTs in the MPU matrix (as shown by the circled regions for MPU/HNTs-5), became broader and the intensity of the reflection increased with increasing HNT loading. Therefore, the crystal structure of HNTs was not altered by their incorporation into the MPU matrix.
 | ||
| Fig. 5 XRD patterns of HNTs, MPU, and MPU/HNTs nanocomposites (raw rubber). | ||
The TEM micrographs of MPU/HNTs nanocomposites reinforced with different wt% HNTs are shown in Fig. 6, from which can be determined the state of dispersion of HNTs in the MPU matrix through melt mixing. The low magnification micrographs show that the HNTs are uniformly distributed throughout the MPU matrix, and that the HNTs demonstrate no significant tendency toward agglomeration or clustering. At high magnification, the individual HNTs are clearly observed at low loading and are well dispersed in the MPU matrix (Fig. 6(B)). However, although the HNTs were significantly dispersed in the MPU matrix at high HNT content, a highly entangled (interconnected) network structure of HNTs is prominently observed in regions of the MPU matrix (Fig. 6(D)). Here, the high viscosity that developed in the high shear internal melt mixing condition hindered the dispersion of HNTs, resulting in the formation of HNTs aggregates within the MPU matrix. Such a preferential distribution further indicates the incorporation of HNTs into the hard segment domains of MPU due to reaction between the OH groups of HNTs and NCO groups of MDI, which also further implies the presence of a strong interfacial interaction between HNTs and the MPU matrix. Individual HNTs with a 15 nm lumen and 30–50 nm external diameter are shown in Fig. 6(E).
 | ||
| Fig. 6 TEM micrographs of ultrathin cryo-cross-sections of MPU/HNTs nanocomposites: (A) and (B) HNTs at 1 wt%:, (C) and (D) HNTs at 5 wt%; (E) HNTs alone. | ||
The phase separation of MPU resulting from thermodynamic incompatibility between the soft and hard segments plays a key role in the physical properties. The glass transition temperature (Tg) of the soft segment molecular chains shift to higher temperatures when the compatibility between soft and hard segments increases and the degree of micro-phase separation decreases. Thus, the degree of phase separation of materials can be determined according to the movement about Tg of the soft segment molecular chains. DMA was employed to measure the tanδ (also referred to as the damping) of the materials, and the results are presented in Fig. 7. Tg was defined as the temperature at which tanδ was a maximum. The Tg values of the MPU/HNTs nanocomposites are greater than that of the pristine MPU, and increased with increasing HNT loading. These results indicate that the HNTs were intensely associated with the hard segments of the MPU, resulting in a lower fraction of hydrogen bonded carbonyl groups in the hard segments and a decrease in the degrees of freedom for the soft segment in the nanocomposites. This also confirms that the HNTs were covalently bonded to the PU molecular chains, resulting in a strong interfacial interaction between HNTs and the hard segments of the MPU, which indicates that incorporating HNTs into the MPU matrix augments its hard segment content. Thus, an increasing Tg was attributed to the strong interfacial interaction between hard microdomains and the HNTs, which made the hard microdomains stiffer. This is particularly evident for 1 and 5 wt% HNT loading, where the Tg of the nanocomposites respectively increased by 10.1 and 23 °C, relative of the Tg of the pristine MPU, which further indicates that the OH groups of HNTs improved compatibility with the MPU. The DMA results are fully consistent with the results of FTIR and XRD analyses.
 | ||
| Fig. 7 Damping factor tanδ of the MPU and MPU/HNTs nanocomposites (raw rubber) as a function of temperature. | ||
The introduction of HNTs as the reinforcing filler in MPU led to improved tensile strength and elongation at break, as summarized in Table 4. The tensile strength was increased from 11.1 MPa for pristine MPU to 23.9 MPa for MPU/HNT-0.5, and further to 24.0 MPa for MPU/HNT-1. Analogously, the elongation at break was increased from 274% for pristine MPU to 389% for MPU/HNT-0.5, and further to 432% for MPU/HNT-1. These results indicate that the HNTs preferentially reinforce the hard segments rather than the soft microdomains of the MPU, contributing toward the large elongation at break of the nanocomposites. These findings were consistent with the results of FTIR and DMA, which implied that the addition of HNTs reinforced the MPU matrix significantly. The reason for the observed reinforcement is because the mechanical properties of nanocomposites mainly depend on the percolated particle networks and interfacial interaction between the polymer matrix and the inorganic filler.48 At low HNT loading (below 5 wt%), HNTs were uniformly dispersed in the MPU nanocomposites, and covalent interactions between HNTs and the hard microdomains of the MPU matrix were formed. To our knowledge, this is the first time such enhancement in mechanical properties has been achieved with only 1 wt% of nanoscale HNTs reinforcement of a raw rubbery matrix.
| Samples | Tensile strength [MPa] | Elongation at break [%] | Young's modulus [MPa] | Hardness [Shore A] |
|---|---|---|---|---|
| MPU | 11.1 | 274 | 13.5 | 85 |
| MPU/HNT-0.5 | 23.9 | 389 | 12.8 | 82 |
| MPU/HNT-1 | 24.0 | 432 | 12.8 | 84 |
| MPU/HNT-5 | 11.0 | 365 | 6.2 | 89 |
As shown in Table 5, Young's modulus and the hardness of the products employing DCP as the curing agent were increased because of the increased crosslinking density. In particular, the Young's modulus of D-MPU/HNT significantly increased with increasing HNT content, attaining the largest value of 48.2 MPa at 5 wt% HNTs, which is more than 7 times greater than that of MPU/HNT-5. Simultaneously, the tensile strength increased from 11.0 to 21.1 MPa. The isocyanate curing process involves a reaction of isocyanate terminal hydroxyl groups with the raw rubber molecules, which expands the raw rubber molecular chain, resulting in an increase in the molecular weight. From Table 6, the Young's modulus and the hardness of the products employing TDID as the curing agent were significantly increased. TDID was added not only to play the role of chain extender, but also as a curing agent to generate branch chains and increase crosslinking density. It is worth mentioning that the Young's modulus of T-MPU/HNT attained a highest value of 101.7 MPa at 5 wt% HNTs, which is more than 16 times greater than that of MPU/HNT-5. Simultaneously, the tensile strength increased from 11.0 to 21.8 MPa. In addition, according to our past experience, the introduction of CaO not only removes water and prevents the formation of bubbles, but can also appropriately increase the hardness of the nanocomposites.
| Samples | Tensile strength [MPa] | Elongation at break [%] | Young's modulus [MPa] | Hardness [Shore A] |
|---|---|---|---|---|
| D-MPU | 10.4 | 272 | 15.2 | 92 |
| D-MPU/HNT-0.5 | 21.7 | 365 | 21.4 | 92 |
| D-MPU/HNT-1 | 22.9 | 347 | 21.5 | 94 |
| D-MPU/HNT-5 | 21.1 | 278 | 48.2 | 97 |
| Samples | Tensile strength [MPa] | Elongation at break [%] | Young's modulus [MPa] | Hardness [Shore A] |
|---|---|---|---|---|
| T-MPU | 16.1 | 206 | 63.9 | 95 |
| T-MPU/HNT-0.5 | 25.8 | 227 | 49.5 | 93 |
| T-MPU/HNT-1 | 21.0 | 264 | 47.6 | 97 |
| T-MPU/HNT-5 | 21.8 | 271 | 101.7 | 99 |
Traditional nanofillers include carbon nanotubes (CNTs), microcrystalline cellulose (MCC), and silicate, and these materials can enhance numerous polymer properties as well as generate improved mechanical properties. Numerous investigations have focused on CNTs, which is technologically demanding to produce in bulk, making it expensive.55 Nanocomposites comprised of multi-wall carbon nanotubes (MWCNT) and PU have been successfully prepared based on hydroxy terminated polybutadiene (HTPB) and MWCNTs prepared according to Shokry et al.56 Mechanical tests have demonstrated that the addition of MWCNTs improved the tensile properties, Young's modulus, and the hardness of the PU matrix without sacrificing the elongation at break. In particular, relative to pristine PU, the addition of 2 wt% pure MWCNTs increased the tensile strength from 6.03 to 11.94 MPa, nearly 2 times greater, the elongation at break was increased from 74.8% to 176.3%, about a 136% increase, and the hardness was increased from 35 A to 44 A. Zahra et al. successfully prepared novel polyurethane/cellulose hybrid bio-nanocomposite films by dispersing MCC in a polyurethane matrix,57 which improved the mechanical properties significantly. Relative to pristine PU, the incorporation of 10 wt% MCC increased the tensile strength of the composites significantly from 34 to 145 MPa, whereas the elongation at break was reduced from 9.8% to 5.8%. Soy polyol-based PU nanocomposites were synthesized with 3-glycidoxypropyltrimethoxysilane modified palygorskite (GPTMSPal) by Wang et al.58 Compared to pristine PU, the GPTMSPal/PU nanocomposite with 12 m% GPTMSPal exhibited a 13.1 °C increase in Tg. In addition, the tensile strength was increased from 3.0 to 12.1 MPa, whereas the elongation at break was reduced from 394.6% to 181.5%. In the present work, HNTs have been shown to offer an inexpensive, low-tech alternative that is morphologically similar to MWCNTs. Moreover, with the addition of only 1 wt% HNTs, we obtained an MPU nanocomposite having a tensile strength of 24.0 MPa, elongation at break of 432%, and a hardness of 84 A, which surpassed those of conventional raw rubbery materials. Therefore, HNTs could replace the more expensive MWCNTs in high-performance polymer nanocomposites.
Conclusions
We have confirmed the synthesis of high tensile strength and elongation at break MPU nanocomposites by adding very small amounts of HNTs as a reinforcing filler. The results from AFM and TEM indicated that HNT fillers of a concentration no greater than 1 wt% were well dispersed within the MPU matrix, and had good adhesion in the interfacial area owing to the reaction between OH groups of HNTs and NCO groups of MDI. A high dispersion state of the HNTs was crucial for improving the mechanical properties. The individual HNTs were covalently bonded with the hard microdomains of the segmented polyether PU, which affected the phase separation of the nanocomposites, and such nanostructure was crucial for stiffening and toughening the MPU without reducing its extensibility, as characterized by FTIR and DMA. With the addition of only 1 wt% HNTs, we obtained an MPU nanocomposite with a tensile strength of 24.0 MPa and an elongation at break of 432%, which surpassed those of conventional raw rubbery materials. Moreover, the addition of reinforcing agents was employed to further enhance the synthetic nanocomposite performance. In particular, the Young's modulus of D-MPU/HNT significantly increased with increasing HNT content, and attained a largest value of 48.2 MPa at 5 wt% HNTs, which is more than 7 times greater than that of MPU/HNT-5. Similarly, the Young's modulus of T-MPU/HNT attained a largest value of 101.7 MPa at 5 wt% HNTs, which is more than 16 times greater than that of MPU/HNT-5. The results of this study are expected to have an impact on widening the practical use and potential applications of HNT-based nanocomposite materials such as hot melt adhesives and TPU.Acknowledgements
The authors thank Ningbo Institute of Industrial Technology and the Instrumental Analysis Center of SJTU for providing equipment to facilitate our AFM and TEM measurements, respectively. We thank LetPub (http://www.letpub.com) for its linguistic assistance during the preparation of this manuscript.References
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52(36), 9422–9441 CrossRef CAS PubMed
.
- H. Sardon, A. C. Engler, J. M. W. Chan, J. M. García, D. J. Coady, A. Pascual, D. Mecerreyes, G. O. Jones, J. E. Rice, H. W. Horn and J. L. Hedrick, J. Am. Chem. Soc., 2013, 135, 16235–16241 CrossRef CAS PubMed
- H. Yeganeh and M. R. Mehdizadeh, Eur. Polym. J., 2004, 40, 1233–1238 CrossRef CAS
- K. Joy, I. K. Mishra and S. H. Chang, Macromol. Rapid Commun., 2003, 24, 671–675 CrossRef
- G. S. Navinchandra, A. S. Harishchandra, D. M. Ananda and S. Mishra, J. Reinf. Plast. Compos., 2013, 32, 935–946 CrossRef
- G. Navinchandra, H. A. Shimpi, A. D. Sonawane and M. S. Mishra, Polym. Bull., 2014, 71, 515–531 CrossRef
- A. K. Barick and Y. W. Chang, High Perform. Polym., 2014, 26, 609–617 CrossRef CAS
- J. Ahenmiller and D. Patterson, Rubber World, 1993, 27 Search PubMed
- R. D. A. Paulmer, C. S. Shah, M. J. Patni and M. V. Pandya, J. Appl. Polym. Sci., 1991, 43, 1953 CrossRef CAS
- N. D. Ghatge and V. B. Phadke, Rubber Age, 1968, 10, 52 Search PubMed
- T. L. Jablonowski and R. J. Scribner, 151st ACS Rubber Division Meeting, Spring, Anaheim Ca., 1997, Paper 61, p. 19.012 Search PubMed
- J. R. Serumgard, 151st ACS Rubber Division Meeting, Spring, Anaheim Ca., 1997, Paper 12, p. 19.012 Search PubMed
- J. Ahnemiller, Rubber Plast. News., 1992, 21(24), 15 Search PubMed
- P. H. McKinstry and M. Chase, Urethanes Technology, 1990, 24–28 Search PubMed
- S. K. N. Kutty and G. B. Nando, Plast. Rubber Process. Appl., 1990, 14, 109 CAS
- H. Shirasaka, US Pat., 6008312, 1999
- A. Singh, US Pat., 4335231, 1982
- Z. S. Petrovic and D. Fajnik, J. Appl. Polym. Sci., 1984, 29, 1031 CrossRef CAS
- R. F. Ziolo, E. P. Giannelis, B. A. Weinstein, M. P. O'Horo, B. N. Granguly, V. Mehrota, M. W. Russell and D. R. Huffman, Science, 1992, 257, 219–222 CrossRef CAS PubMed
- H. Gleiter, Adv. Mater., 1992, 4, 474–481 CrossRef CAS
- S. J. Komarneni, Mater. Chem., 1992, 2, 1219–1230 RSC
- B. M. Novak, Adv. Mater., 1993, 5, 422–433 CrossRef CAS
- R. S. Sinha and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. D. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80–83 CrossRef CAS PubMed
- S. M. Liff, N. Kumar and G. H. McKinley, Nat. Mater., 2007, 6, 76–83 CrossRef CAS PubMed
- D. Y. Cai, K. Yusoh and M. Song, The mechanical properties and morphology of a graphite oxide nanoplatelet/polyurethane composite, Nanotechnology, 2009, 20, 685–712 Search PubMed
- B. S. Hsiao, H. White, M. Rafailovich, P. T. Mather, H. G. Jeon, S. Phillips, J. Lichtenhan and J. Schwab, Polym. Int., 2000, 49, 437–440 CrossRef
- H. Liu, J. Xu, Y. Li, B. Li, J. Ma and X. Zhang, Macromol. Rapid Commun., 2006, 27, 1603–1607 CrossRef CAS
- R. Sen, B. Zhao, D. Perea, M. E. Itkis, H. Hu, J. Love, E. Bekyarova and R. C. Haddon, Nano Lett., 2004, 4, 459–464 CrossRef CAS
- H. Koerner, W. D. Liu, M. Alexander, P. Mirau, H. Dowty and R. A. Vaia, Polymer, 2005, 46, 4405–4420 CrossRef CAS
- H. S. Xia and M. Song, Soft Matter, 2005, 1, 386–394 RSC
- J. Kwon and H. J. Kim, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3973–3985 CrossRef CAS
- S. M. Liff, N. Kumar and G. H. McKinley, Nat. Mater., 2007, 6, 76–83 CrossRef CAS PubMed
- Z. Wang and T. J. Pinnavaia, Chem. Mater., 1998, 10, 3769–3771 CrossRef CAS
- C. Zilg, R. Thomann, R. Mulhaupt and J. Finter, Adv. Mater., 1999, 11, 49–52 CrossRef CAS
- Y. I. Tien and K. H. Wei, Macromolecules, 2001, 34, 9045–9052 CrossRef CAS
- B. Finnigan, K. Jack, K. Campbell, P. Halley, R. Truss, P. Casey, D. Cookson, S. King and D. Martin, Macromolecules, 2005, 38, 7386–7396 CrossRef CAS
- E. Joussein, S. Petit, J. Churchman, B. Theng, D. Righi and B. Delvaux, Clay Miner., 2005, 40, 383–426 CrossRef CAS
- B. Singh, Clays Clay Miner., 1996, 44, 191–196 CrossRef CAS
- R. L. Frost and H. F. Shurvell, Clays Clay Miner., 1997, 45, 68–72 CrossRef
- M. Du, B. Guo, X. Cai, Z. Jia, M. Liu and D. Jia, Morphology and properties of halloysite nanotubes reinforced polypropylene nanocomposites, e-Polymers, 2008, 130, 1–14 Search PubMed
- K. M. Ng, Y. T. R. Lau, C. M. Chan, L. T. Weng and J. Wu, Surf. Interface Anal., 2011, 43, 795–802 CrossRef CAS
- H. Ismail, S. Z. Salleh and Z. Ahmad, Mater. Des., 2013, 50, 790–797 CrossRef CAS
- D. Tao, Y. J. Higaki and W. Ma, Polymer, 2015, 60, 284–291 CrossRef CAS
- M. X. Liu, Z. X. Jia, D. M. Jia and C. R. Zhou, Prog. Polym. Sci., 2014, 39, 1498–1525 CrossRef CAS
- L. Jiang, C. Zhang, M. K. Liu, Z. Yang, W. W. Tjiu and T. X. Liu, Compos. Sci. Technol., 2014, 91, 98–103 CrossRef CAS
- B. Lecouvet, M. Sclavons, S. Bourbigot, J. Devaux and C. Bailly, Polymer, 2011, 52, 4284–4295 CrossRef CAS
- A. Pei, J. M. Malho, J. Ruokolainen, Q. Zhou and L. A. Berglund, Macromolecules, 2011, 44, 4422–4427 CrossRef CAS
- C. Bueno-Ferrer, E. Hablot, M. C. Garrigos, S. Bocchini, L. Averous and A. Jimenez, Polym. Degrad. Stab., 2012, 97, 1964–1969 CrossRef CAS
- E. Hablot, D. Zheng, M. Bouquey and L. Averous, Macromol. Mater. Eng., 2008, 293, 922–929 CrossRef CAS
- L. Hojabri, X. Kong and S. S. Narine, Biomacromolecules, 2009, 10, 884–891 CrossRef CAS PubMed
- A. O. Sanches, L. H. S. Ricco, L. F. Malmonge, M. J. Silva, W. K. Sakamoto and J. A. Malmonge, Polym. Test., 2014, 40, 99–105 CrossRef CAS
- M. Song, H. S. Xia, K. J. Yao and D. J. Hourston, Eur. Polym. J., 2005, 41, 259–266 CrossRef CAS
- J. Marini and R. Bretas, Polym. Eng. Sci., 2013, 53, 1512–1528 Search PubMed
- M. X. Liu, Z. X. Jia, D. M. Jia and C. R. Zhou, Prog. Polym. Sci., 2014, 39, 1498–1525 CrossRef CAS
- S. A. Shokry, A. K. EI Morsi, D. M. Jia, M. S. Sabaa, R. R. Mohamed and H. E. EI Sorogy, Egypt. J. Pet., 2015, 24, 145–154 CrossRef
- Z. Rafiee and V. Keshavarz, Prog. Org. Coat., 2015, 86, 190–193 CrossRef CAS
- C. S. Wang, Q. S. Wu, F. Liu, J. An, R. Lu, H. F. Xie and R. S. Cheng, Appl. Clay Sci., 2014, 101, 246–252 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2015 |