
Mechanism of Pd-catalyzed C(sp3)–H activation of aliphatic amines: an insight from DFT calculations†
Abstract
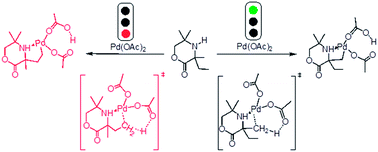
A theoretical understanding of the Pd-catalyzed C(sp3)–H activation of aliphatic amines has been examined using the B3LYP density functional theory. The concerted metalation–deprotonation (CMD) mechanism is identified in the rate-determining steps of all possible reaction pathways. The rate- and regio-determining step of the catalytic cycle is deprotonation of the Cmethyl–H bond through a six-membered cyclopalladation transition state. According to the relative activation barriers, the Cmethyl–H activation is kinetically and thermodynamically more favorable than the Cethyl–H activation. More important, the only acetoxylation product is located, ignoring the diethyl-substituted or the dimethyl-substituted in the morpholine and not producing the lactone amines molecules, which is in good agreement with the experimental observations.
 Please wait while we load your content...
Please wait while we load your content...

