
Bulk solvent extraction of biomass slurries using a lipid trap†
Abstract
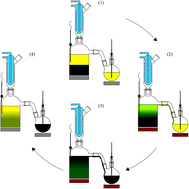
Extraction of lipids and hydrophobic metabolites from microbial sources remains an obstacle in the production of these compounds at the laboratory and industrial scale. Analytical techniques for the total extraction of non-polar metabolites from biological material are well established, but rely on expensive and time consuming processes. This makes these techniques unsuitable for direct translation to continuous or large volume systems, unable to move beyond proof-of-concept studies, and leaves a major gap in the translation of new bio-products requiring a purified extract. Here we attempt to bridge that gap by demonstrating the use of a semi-continuous liquid–liquid extraction system capable of bulk lipid extraction from wet, untreated biomass, and simultaneous concentration of the unmodified extract in a lipid trap. A 1.8 L model was used to evaluate system dynamics with bacterial, fungal, algal, and plant feedstock, prior to scaling the system by an order of magnitude to demonstrate large-scale viability. Extraction efficiency was above 90% for each feedstock compared to standard Bligh and Dyer extraction. Following scale-up, extraction was performed on upwards of 4 kg of slurry (660 g dry weight), yielding an average efficiency of 96%, and allowing generation of a crude extract at a scale not previously possible in a laboratory setting. The resulting system allows for direct and high-throughput extraction of biomass sources without pretreatment, specialized instrumentation, or intensive user input.
 Please wait while we load your content...
Please wait while we load your content...

