
Retracted Article: A rapid and novel method for the synthesis of 5-substituted 1H-tetrazole catalyzed by exceptional reusable monodisperse Pt NPs@AC under the microwave irradiation†
Esma Erken‡
a,
İbrahim Esirden‡b,
Muharrem Kaya*a and
Fatih Sen*a
aBiochemistry Department, Faculty of Arts and Science, Dumlupınar University, Evliya Çelebi Campus, 43100 Kütahya, Turkey. E-mail: fatih.sen@dpu.edu.tr
bChemistry Department, Faculty of Arts and Science, Dumlupınar University, Evliya Çelebi Campus, 43100 Kütahya, Turkey
First published on 7th August 2015
Abstract
A series of 5-substituted 1H-tetrazoles were synthesized in DMF by the [3 + 2] cycloaddition reaction under the effect of microwave irradiation (10–30 min, fixed mode, 90 °C, 140 W) in the presence of highly efficient superior catalyst. For this reaction, different aromatic nitriles with the sodium azide were used and superior monodisperse (Md) platinum nanoparticles (Pt NPs) decorated on activated carbon (AC) served as a catalyst. Md-Pt NPs@AC were reproducibly and easily produced by double solvent reduction of PtCl4 in room temperature and characterized by transmission electron microscopy (TEM), the high resolution electron micrograph (HRTEM), X-ray diffraction (XRD), atomic force microscopy (AFM) and X-ray photoelectron spectroscopy (XPS). The sum of their results shows the formation of highly crystalline and colloidally stable Md-Pt NPs@AC. The catalytic performance of these new NPs were investigated for the synthesis of 5-substituted 1H-tetrazoles, in which they were found to be exceptional reusable, isolable, stable and highly efficient heterogeneous catalyst. All prepared tetrazole products were obtained with perfect yield by using current heterogeneous catalyst and characterized by melting points, FT-IR, 1H-NMR, 13C-NMR and HRMS analyzes.
Introduction
Tetrazoles have been captivating increasing attention, due to their wide application, such as they show strong activities in pharmaceutical chemistry as anticancer,1 antimicrobial,2–4 analgesics,5 antibacterial,6 antifungal,7 and antihypertensive agents.8 Additionally, these nitrogen-rich ring systems have applications in material science including photography,9 propellants10 and explosives.11 The significance of tetrazole and its derivatives has enhanced because of their use as an isosteric replacement for carboxylic acids in medicinal chemistry.12 The pKa of tetrazole is roughly 4.90 and similar to that of –CO2H.13 The angiotensin II receptor antagonist, losartan, is one example where tetrazole is being used in replacement of –CO2H.14 The most substantial drugs containing a tetrazole ring are antihypertensive drugs, losartan (Scheme 1).15 | ||
| Scheme 1 The chemical structure of losartan. | ||
Even though tetrazoles were explored more than 130 years ago, research of these compounds was started in the latter half of the 20th century. Tetrazole was first prepared by the reaction of the hydrogen cyanide and the dehydrated hydrazoic acid in difficult conditions. The reactants used in the essential method are water-reactive and highly toxic. Especially, the hydrazoic acid is explosive, volitant and extremely toxic as well. In view of these disadvantages, there has been an upsurge to change the methods of the preparation of tetrazoles and several methods have been informed for the synthesis of 5-substituted 1H-tetrazoles. Most of them are based on the addition of the nitrile group of the trimethylsilyl azide (TMSN3) and the sodium azide (NaN3).16–21 Also, different new pathways have been improved,22–26 such as the catalytic method using a stoichiometric amount of inorganic salts27–32 and metal complex,33 also using TMSN3 and TBAF instead of metal salts under solvent-free conditions17 or in micellar media34 and ionic liquid.35 However, these homogeneous catalysts pose difficulties in reusability, recovery and separation. For this reason, the heterogeneous alternatives are highly attractive and have already captivated much attention. Due to the heterogeneous catalyst may be recovered and reusable, recently several heterogeneous catalytic systems were reported using, for example, nanocrystalline ZnO,36 Fe3O4@SiO2/salen Cu(II),37 ZnO nanoflakes,38 Cu2O39 and ZnO/Co3O4 (ref. 40) as catalysts. Since the problem is low reusability performance and low yield in most of those catalytic systems.
Herein we report Md-Pt NPs@AC as a new heterogeneous catalyst for synthesis of tetrazoles with superior catalytic activity. The current Md-Pt NPs@AC have been used for the first time for these types of synthesis reactions. In addition, the current catalyst has features such as easy separation, recovery and reusability. Moreover, in recent years, a simplified procedure for the synthesis of tetrazole compound is formed by passing the modern microwave systems due to high reflux time in the reactions.19,41–43 The most important advantages of using microwave technology in organic syntheses are short reaction time, high yield and purity. Moreover, Palde and Jamison have synthesized tetrazoles under continuous-flow microreactor system with high yield by using homogenous catalysts.44 Mukhopadhyay et al. have also synthesized mono- and bis-tetrazolato complexes via 1,3-dipolar cycloaddition of organonitriles with homogenous platinum(II)-bound azides as catalyst under MW conditions.45 However, as mentioned before, these homogeneous catalysts pose difficulties in reusability, recovery and separation. For this reason, addressed herein, we have used Md-Pt NPs@AC as an effective heterogeneous catalyst system under microwave (MW) irradiation method instead of the traditional reflux method in order to describe a rapid, appropriate, attractive, useful and simple method for the synthesis of 5-substituted 1H-tetrazole derivatives from the reaction of different aromatic nitriles with sodium azide in DMF.
Experimental methods
The preparation of Md-Pt NPs@AC
Pt NPs was synthesized by double solvent reduction method. Summarizing this method, ethanol and superhydride were used to reduce the mixture of 0.25 mmol of PtCl4 dissolved in small amount of dehydrated tetrahydrofuran and 0.25 mmol of 1-aminopropane ligand. The observation of a brown–black colour in the solution indicates the formation of amine-stabilized Pt NPs. Lastly, in the room temperature, the solid Pt NPs was dried under vacuum. The prepared platinum nanoparticles were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio with activated carbon (AC), which is used as a support.46a–f
:10 ratio with activated carbon (AC), which is used as a support.46a–f
General procedure for preparation of tetrazole derivatives 3–12 (Table 2) using Md-Pt NPs@AC as the catalyst
A mixture of nitrile (0.3 mmol), sodium azide (0.45 mmol), Md-Pt NPs@AC (7 mg) and DMF (1.5 mL) were added in an oven-dried 10 mL vessel. Then, the vessel was placed in microwave and irradiated at 140 W power and 90 °C for 10–30 min in fixed mode. The progress of the reaction was monitored by thin layer chromatography. After completion, the reaction mixture was cooled to room temperature and the catalyst was separated from the media by the help of centrifugation method. Reaction mixture was treated with 5 mL HCl (4 mol L−1). The resultant organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 × 15 mL). The extracted ethyl acetate was concentrated under the reduced pressure. The combined product was recrystallized from acetic acid to obtain pure 5-phenyl 1H-tetrazole as a white powder, Mp: 215–217 °C (lit. 215–216 °C),47 FT-IR (cm−1): 3053, 2979, 2906, 2833, 2682, 2601, 1607, 1561, 1285, 1254 1H NMR (300 MHz, DMSO-d6): δ = 7.60–7.65 (m, 3H, Ar–H), 8.03–8.07 (m, 2H, Ar–H), 16.85 (br, 1H, –NH) ppm, 13C NMR (75 MHz, DMSO-d6): δ = 124.60, 127.40, 129.79, 131.62, 155.77 ppm, HRMS (QTOF-ESI): m/z [M − H]− calcd for C7H5N4: 145.0514; found [M − H]−: 145.0517.Results and discussion
The preliminary characterization of Md-Pt NPs@AC was performed by XRD, TEM, HRTEM, AFM and XPS spectroscopies.X-ray diffraction (XRD) pattern of Md-Pt NPs@AC are illustrated in Fig. 1. 2θ = 39.87, 46.21, 67.67, 81.47 and 85.96 diffraction peaks are due to Pt (111), (200), (220), (311), (320) planes of the face-centered cubic (fcc) crystal lattice of platinum, respectively. (JCPDS-ICDD, card no. 04-802), the Pt (220) diffraction peak of the prepared Md-Pt NPs@AC is used to calculate the lattice parameter values and average crystallite size of the metal particles. The lattice parameter value of the resulting Md-Pt NPs@AC were calculated as 3.921 Å by Pt (220) diffraction peak from the following equation.48
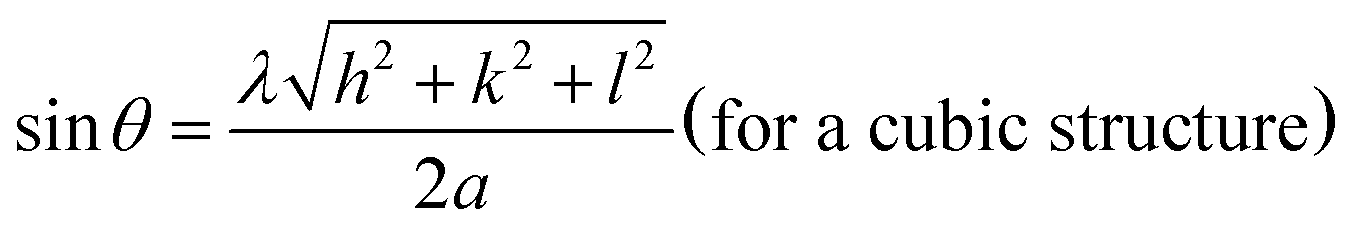
 | ||
| Fig. 1 XRD of Md-Pt NPs@AC. | ||
Furthermore, the average crystallite platinum particle size of the prepared catalyst was calculated to be about 3.56 ± 0.47 nm using following Scherrer equation with XRD as Pt (220);49

TEM image (>100 particles were counted) of Md-Pt NPs@AC are given in Fig. 2, they show the entity of Md-Pt NPs@AC in the range of 3.2–4.1 nm with an average diameter of 3.63 ± 0.42 nm. In addition, the high resolution electron micrograph (HRTEM) of the catalyst is shown in Fig. 2. HRTEM results indicated that most of particles are in spherical shape, and no agglomerations are observed in our catalyst. Fig. 2 also displays the representative atomic lattice fringes obtained by high resolution transmission electron microscopy for Md-Pt NPs@AC. Moreover, the chosen area HRTEM image of Md-Pt NPs@AC displays the crystalline speciality of these Md-Pt NPs@AC with a crystalline spacing of 0.228 nm which is exactly same with nominal Pt (111) spacing of 0.228 nm.50–52
 | ||
| Fig. 2 High resolution transition electron micrograph and particle size histogram of Md-Pt NPs@AC. | ||
Atomic force microscopy (AFM) was used to see the height diameter distributions which are shown in Fig. 3. The height values of Md-Pt NPs@AC (3.61 ± 0.74) are in good agreement with TEM and XRD results.
 | ||
| Fig. 3 General surface topography of Md-Pt NPs@AC and histogram of height of particles obtained from AFM data. | ||
X-ray photoelectron spectroscopy (XPS) was used to designate the surface compositions and chemical oxidation states of Pt in the nanocatalysts. To that end, the Pt 4f region of spectrum was analyzed and the fitting of XPS peak was performed by Gaussian–Lorentzian method and the relative intensity of the species was estimated by the integral of each peak. Exact binding energies (±0.3 eV) were decided by referencing to the C 1s peak at 284.6 eV at XPS spectrum. Fig. 4 shows the Pt 4f photoelectron spectrum which comprise of two pairs of doublet. The most intense doublet at about 71.0 and 74.3 eV is a signature of metallic platinum53,54 and the other doublet at about 74.5 and 77.8 eV is most likely by very small fraction of oxidized Pt4+ species most probably due to unreduced Pt precursor or PtOx species formed along the catalyst exposing to the atmosphere as shown in Table 1. The ratios of Pt(0) to Pt(IV) for the prepared catalysts were calculated from the relative peak area of the Pt 4f spectrum.
 | ||
| Fig. 4 Pt 4f electron spectra of Md-Pt NPs@AC. | ||
| Pt 4f7/2 | Pt 4f7/2 | Pt(0)/Pt(IV) | |
|---|---|---|---|
| Pt(0) | Pt(IV) | ||
| Md-Pt NPs@AC | 71.0 (84.5) | 74.3 (15.5) | 5.45 |
It is worth mentioning that Pt NPs is often used as a catalyst in alcohol oxidation, dehydrogenation and/or hydrolysis reactions45,47,50 but its use as a catalyst for synthesizing 5-substituted 1H-tetrazoles from sodium azide and nitriles has not been reported before. Furthermore, the usage of microwave irradiation method in current work, may be regarded as a rapid, appropriate, attractive, useful, simple and one of the safest procedures for the synthesis of 5-substituted 1H-tetrazole derivatives from the reaction of different aromatic nitriles with sodium azide in DMF using Md-Pt NPs@AC as an effective heterogeneous catalyst system.
The general synthesis method shown in Scheme 2 was employed to prepare the tetrazole compounds (Table 2, entries 3–12). Tetrazole products were prepared by means of a one-pot reaction producing high yields and providing a simple work-up procedure.
 | ||
| Scheme 2 Synthesis of 5-substituted 1H-tetrazole compounds. | ||
| Entry | Product | Time (min)/effect (W) | Yieldb (%) | TON c | TOFd (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: benzonitrile (0.3 mmol), NaN3 (0.45 mmol), Md-Pt NPs@AC (7 mg), DMF (1.5 mL), reaction time 20 min, temp. 90 °C and effect 140 W.b Isolated yields.c TON = moles of product formed per mole catalyst.d TOF = TON/time (h) of the reaction. | |||||
| 3 |  |
20/140 | 95 | 7.92 | 23.76 |
| 4 |  |
12/140 | 97 | 8.08 | 40.40 |
| 5 |  |
15/140 | 96 | 7.99 | 31.96 |
| 6 |  |
15/140 | 98 | 8.17 | 32.68 |
| 7 |  |
18/140 | 94 | 7.83 | 26.10 |
| 8 | 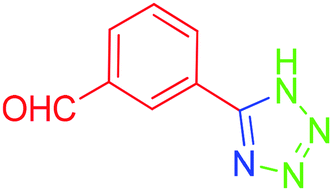 |
18/140 | 93 | 7.75 | 25.83 |
| 9 | 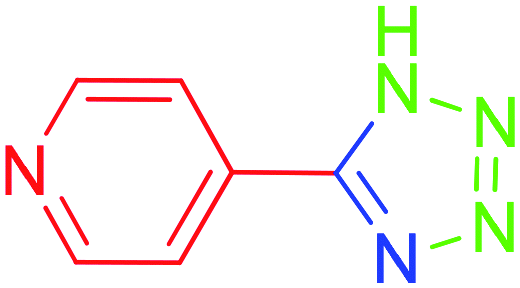 |
10/140 | 99 | 8.25 | 49.50 |
| 10 | 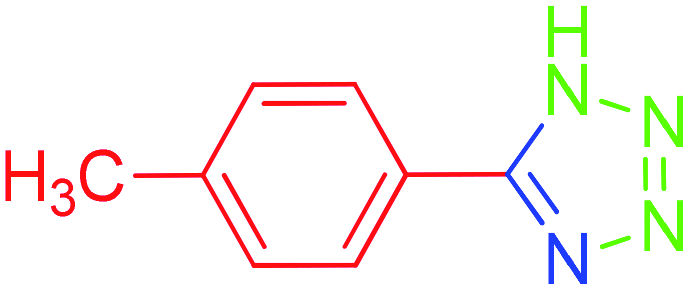 |
25/140 | 92 | 7.67 | 18.41 |
| 11 |  |
30/140 | 90 | 7.50 | 15.00 |
| 12 |  |
30/140 | 89 | 7.41 | 14.82 |
Aromatic nitriles 1 were transformed into tetrazoles with the sodium azide 2 in the molar ratio of 1:1.5 using Md-Pt NPs@AC as the catalyst under the effect of microwave irradiation. By changing the nature of the substituents present in the aromatic nitrile components, a rather large chemical diversity can be incorporated in tetrazoles (Scheme 2, Table 2). These several substituents are subsumed in the compounds of nitro, aldehyde, halogen (Cl, Br), pyridine moiety, methyl and acetanilide (Table 2).
We investigated the effects of the solvent on the model reaction (Table 3, entries 1–6). The model reaction was performed in DMF, DMSO, ethanol, toluene, THF and water. The reactions gave poor yields when water, THF, ethanol and toluene were used as solvents. For this reason, these solvents were determined not to be suitable for this model reaction. Although the phenyl tetrazole was obtained in a good yield but a long reaction time in DMSO, the one in DMF was performed in a short reaction time with the best yield. Therefore, it can be thought that DMF is a superior solvent compared to the others.
| Entry | Solvent | Temperature (°C) | Time (min)/effect (W) | Yield (%) |
|---|---|---|---|---|
| 1 | DMF | 90 | 20/140 | 95 |
| 2 | DMSO | 120 | 30/140 | 87 |
| 3 | Ethanol | 80 | 40/140 | 72 |
| 4 | Toluene | 110 | 35/140 | 54 |
| 5 | THF | 70 | 40/140 | 47 |
| 6 | Water | 100 | 45/140 | 20 |
The concentration of the Md-Pt NPs@AC plays a crucial role in the success of the reactions in terms of the rate and the yields, and these experiments are summarized in Table 4. In the absence of catalyst the phenyl tetrazole was obtained 0% yield in 50 °C (45 min), ≤10% yield in 70 °C and 18% yield in 90 °C (45 min). Increasing the catalyst to 4, 6, 7, 8 and 9 mg resulted in increasing the reaction yields to 80, 88, 95, 95 and 95% the reaction times varied from 10 to 45 minutes. The phenyl tetrazole product was obtained in the model reaction in the presence of 7 mg Md-Pt NPs@AC in DMF in a high yield (95%) and very short time (20 min). Use of just 7 mg Md-Pt NPs@AC in DMF is enough to afford an efficient synthesis. In conclusion, the best catalyst amount was determined to be 7 mg Md-Pt NPs@AC (Table 4, entry 6) in current work.
| Entry | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Catalyst (mg) | None | None | None | 4 | 6 | 7 | 8 | 9 |
| Time (min) | 45 | 45 | 45 | 45 | 30 | 20 | 15 | 10 |
| Temperature (°C) | 50 | 70 | 90 | 90 | 90 | 90 | 90 | 90 |
| Effect (W) | 140 | 140 | 140 | 140 | 140 | 140 | 140 | 140 |
| Yield (%) | — | ≤10 | 18 | 80 | 88 | 95 | 95 | 95 |
Table 5 indicates the comparison of the activity of the different heterogeneous catalysts with that of prepared Md-Pt NPs@AC by considering the yield for the reaction. To the best of our knowledge, the current Md-Pt NPs@AC catalyst gives the best catalytic activities among the other catalysts in literature such as nanocrystalline ZnO, ZnO nanoflakes, montmorillonite K-10 clay, nano CSMIL, CuFe2O4 nanoparticles and mesoporous ZnS. Md-Pt NPs@AC authenticated to be a better catalyst than other catalysts with respect to higher reusability and very low reaction time performances. The prepared Md-Pt NPs@AC have been also compared with PtCl4 which is the precursor material of Md-Pt NPs@AC as shown in Table 2. When we have used PtCl4 as a precatalyst and Md-Pt NPs@AC as a catalyst under the same conditions for the preparation of 5-substituted 1H-tetrazoles, we observed 79% and 95% yield, respectively (Table 2). This case can be explained that the sole stabilizer present in PtCl4 system is the weakly coordinating chloride anion cannot obtain enough stabilization for the preparation of 5-substituted 1H-tetrazoles. During the presence of capturing ligand and AC, the Md-Pt NPs@AC are found to be highly stable towards agglomeration over months. It shows that capturing ligand and AC are a powerful stabilizing and supporting agent for the Md-Pt NPs@AC. After the washing of the Md-Pt NPs@AC with dry ethanol for removing of excess ligand, they can be isolated readily as solid by vaporization of solvent under vacuum atmosphere.
| Entry | Catalyst | Time | Temperature (°C) | TON | TOF | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Yield after third cycle.b Yield after second cycle.c Yield after fifth cycle.d Yield after sixth cycle. | |||||||
| 1 | Nanocrystalline ZnO | 14 h | 120 (Thermal) | 1.54 | 0.11 | 72, 66a | 36 |
| 2 | ZnO nanoflakes | 14 h | 125 (thermal) | 1.77 | 0.13 | 87 | 38 |
| 3 | Montmorillonite K-10 clay | 16 min | 130 (MW) | — | — | 75, 56b | 41 |
| 4 | Nano CSMIL | 7 h | 120 (thermal) | — | — | 87 | 55 |
| 5 | CuFe2O4 nanoparticles | 12 h | 120 (thermal) | 2.05 | 0.17 | 82, 75c | 56 |
| 6 | Mesoporous ZnS | 36 h | 120 (thermal) | 2.15 | 0.06 | 86 | 57 |
| 7 | PtCl4 | 20 min | 90 (MW) | — | — | 79 | This work |
| 8 | Md-Pt NPs@AC | 10 h | 120 (thermal) | 7.50 | 0.75 | 90 | This work |
| 9 | Md-Pt NPs@AC | 20 min | 90 (MW) | 7.92 | 23.76 | 95, 88d | This work |
We have also discussed the comparison of the activities of Md-Pt NPs@AC with and without (thermal) MW irradiation and showed that Md-Pt NPs@AC has better catalytic activity under MW compared to the thermal conditions as shown in Table 5.
To understand the scope and effectiveness of the catalyst Md-Pt NPs@AC, different substituents benzonitriles were selected and used in the determined conditions. These [3 + 2] cycloaddition reactions play a vital role on the nitrile compound activity. 5-Substituted 1H-tetrazoles were obtained in a shorter time with very good yields than electron withdrawing functional groups containing benzonitriles (Table 2, entries 4–8) and heteroaromatic nitriles compound such as 4-pyridinecarbonitrile (Table 2, entry 9) while benzonitriles containing electron-releasing groups (Table 2, entry 10–12) converted to tetrazoles in longer reaction times. For example, –NO2, –Br, –Cl and –CHO, which contain electron-withdrawing groups, react faster in better yields when compared to the nitriles containing electron-releasing groups like –CH3 and –NHAc. In fact, 4-pyridinecarbonitrile was transformed into the tetrazole compound in a much shorter period of time with a very good yield. All the products were characterized by melting points, FT-IR, NMR, and HRMS analyses.
The FT-IR results indicated that the disappearance of strong and sharp absorption –CN band, and the appearance of –NH bands in the 2500–3000 cm−1, proved the formation of 5-substituted 1H-tetrazoles. Besides, the FT-IR spectra of all the products show bands at 1515–1606 cm−1 (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and 1233–1293 cm−1 (N–NN–). Analysing the 13C NMR spectra of all the products, signals at 150–160 ppm is correspond to the quaternary carbon of tetrazole ring (NH–CN). In addition, analysing the HRMS spectra of all the products observation of belonging to the proposed structure [M − H]− peak and then the loss of 28 units mass that resulted from the loss of N2 from a tetrazole ring supports the tetrazole formation (Fig. S1–S7†).
N) and 1233–1293 cm−1 (N–NN–). Analysing the 13C NMR spectra of all the products, signals at 150–160 ppm is correspond to the quaternary carbon of tetrazole ring (NH–CN). In addition, analysing the HRMS spectra of all the products observation of belonging to the proposed structure [M − H]− peak and then the loss of 28 units mass that resulted from the loss of N2 from a tetrazole ring supports the tetrazole formation (Fig. S1–S7†).
The model reaction (benzonitrile, NaN3 in a 1:1.5 molar ratio in DMF) was implemented successfully within 20 min in the presence of 7 mg heterogeneous Md-Pt NPs@AC in the first run. Md-Pt NPs@AC, which recovered by centrifuges after each reaction, can be reutilized for 6 consecutive times in the new experiments without influential yield loss and produce products with purities similar to those achieved in the first run (Fig. 5). As a conclusion, the reusability performances of the prepared Md-Pt NPs@AC have been compared with the others reported in literature and it was found that our Md-Pt NPs@AC have the best reusability performances to the best of our knowledge as shown in Fig. 5.
 | ||
| Fig. 5 Reusability performance of Md-Pt NPs@AC. | ||
Conclusions
Easy, effective and practical synthetic method has been developed for the synthesis of 5-substituted 1H-tetrazoles via the successive [3 + 2] cycloaddition reactions of the various aryl nitrile compounds and sodium azide in the presence of novel, stable, exceptional reusable, isolable, bottleable, long-lived and highly efficient Md-Pt NPs@AC as the catalyst. This nano sized heterogeneous catalyst showed the best catalytic activities among the other catalysts in literature for the synthesis of 5-substituted 1H-tetrazoles most likely due to low crystalline particle size, high chemical surface area and high % Pt(0) contents of the catalyst. Besides, this reusable catalyst offers other advantages like simple work-up and high yields. Furthermore, the given methodology is efficient and environmentally benign. As indicated this work, the yields of the compounds prepared by microwave irradiation method are viably higher than those reported under traditional conditions in the literature. The current method and catalyst would be a rather attractive synthetic method for the future works.Acknowledgements
This research was financed by Dumlupınar University Research Fund (Grant No. 2014-05 and 2014-62). The partial support by the Science Academy is gratefully acknowledged.Notes and references
- C. N. S. S. Pavan Kumar, D. K. Parida, A. Santhoshi, A. K. Kota, B. Sridhar and V. J. Rao, Med. Chem. Commun., 2011, 2, 486–492 RSC.
- Y. Yıldırır, M. F. Us, N. Çolak, H. Özkan, S. Yavuz, A. Disli, S. Ozturk and L. Turker, Med. Chem. Res., 2009, 18, 91–97 CrossRef.
- A. Dişli, S. Mercan and S. Yavuz, J. Heterocycl. Chem., 2013, 50, 1446 CrossRef PubMed.
- G. D. Çelik, A. Disli, Y. Oner and L. Acik, Med. Chem. Res., 2013, 22, 1470–1479 CrossRef.
- S. Yavuz, Y. Ünal, Ö. Pamir, D. Yılmazer, Ö. Kurtipek, M. Kavutçu, M. Arslan, M. Ark and Y. Yıldırır, Arch. Pharm. Chem. Life Sci., 2013, 346, 455–462 CrossRef CAS PubMed.
- T. Narasaiaha, D. S. Raoa, S. Rasheeda, G. Madhavaa, D. Srinivasulua, P. B. Naidub and C. N. Rajua, Pharm. Lett., 2012, 4, 854–862 Search PubMed.
- A. M. Malik, S. A. Al-Thabaiti and M. A. Malik, Int. J. Mol. Sci., 2012, 13, 10880–10898 CrossRef PubMed.
- E. P. Ellis and G. B. West, Progress in Medicinal Chemistry, Biomedical Press, 1980, vol. 17, p. 151 Search PubMed.
- L. M. T. Frija, A. Ismael and M. L. S. Cristiano, Molecules, 2010, 15, 3757–3774 CrossRef CAS PubMed.
- R. Damavarapu, T. M. Klapötke, J. Stierstorfer and K. R. Tarantik, Propellants, Explos., Pyrotech., 2010, 35, 395–406 CrossRef CAS PubMed.
- C. Zhaoxu and X. Heming, Propellants, Explos., Pyrotech., 1999, 24, 319–324 CrossRef.
- R. Herr, Bioorg. Med. Chem., 2002, 10, 3379 CrossRef CAS.
- L. D. Hansen, E. J. Baca and P. S. Scheiner, J. Heterocycl. Chem., 1970, 7, 991 CrossRef CAS PubMed.
- B. C. Bookser, Tetrahedron Lett., 2000, 41, 2805–2809 CrossRef CAS.
- R. R. Wexler, W. J. Greenlee, J. D. Irvin, M. R. Goldberg, K. Prendergast, R. D. Smith and P. B. Timmermans, J. Med. Chem., 1996, 39, 625 CrossRef CAS PubMed.
- Y. Yildirir, Ö. Pamir, S. Yavuz and A. Dişli, J. Heterocycl. Chem., 2013, 50, 93 CrossRef PubMed.
- D. Amantini, R. Beleggia, F. Fringuelli, F. Pizzo and L. Vaccoro, J. Org. Chem., 2004, 69, 2896–2898 CrossRef CAS PubMed.
- W. K. Su, Z. Hong, W. G. Shan and X. X. Zhang, Eur. J. Org. Chem., 2006, 12, 2723–2726 CrossRef PubMed.
- J. J. Shie and J. M. Fang, J. Org. Chem., 2007, 72, 3141–3144 CrossRef CAS PubMed.
- S. Hajra, D. Sinha and M. Bhowmick, J. Org. Chem., 2007, 72, 1852–1855 CrossRef CAS PubMed.
- J. M. Keith, J. Org. Chem., 2006, 71, 9540–9543 CrossRef CAS PubMed.
- V. Aureggi and G. Sedelmeier, Angew. Chem., Int. Ed., 2007, 119, 8592 CrossRef PubMed.
- D. P. Curran, S. Hadida and S. Y. Kim, Tetrahedron, 1999, 55, 8997 CrossRef CAS.
- S. J. Wittenberger and B. G. Donner, J. Org. Chem., 1993, 58, 4139–4141 CrossRef CAS.
- B. E. Huff and M. A. Staszak, Tetrahedron Lett., 1993, 34, 8011–8014 CrossRef CAS.
- J. V. Duncia, M. E. Pierce and J. B. Santella III, J. Org. Chem., 1991, 56, 2395–2400 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210–12216 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, Org. Lett., 2002, 4, 2525–2527 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed.
- J. Bonnamour and C. Bolm, Chem.–Eur. J., 2009, 15, 4543–4545 CrossRef CAS PubMed.
- İ. Esirden, E. Başar and M. Kaya, Chem. Pap., 2015, 69, 1231–1236 Search PubMed.
- L. Bosch and J. Vilarrasa, Angew. Chem., Int. Ed., 2007, 46, 3926–3930 CrossRef CAS PubMed.
- B. S. Jursic and B. W. LeBlanc, J. Heterocycl. Chem., 1998, 35, 405–408 CrossRef CAS PubMed.
- B. Schmidt, D. Meid and D. Kieser, Tetrahedron, 2007, 63, 492–496 CrossRef CAS PubMed.
- M. L. Kantam, K. B. S. Kumar and C. Sridhar, Adv. Synth. Catal., 2005, 347, 1212–1214 CrossRef CAS PubMed.
- F. Dehghani, A. R. Sardarian and M. Esmaeilpour, J. Organomet. Chem., 2013, 743, 87–96 CrossRef CAS PubMed.
- A. Sinhamahapatra, A. K. Giri, P. Pal, S. K. Pahari, H. C. Bajaj and A. B. Panda, J. Mater. Chem., 2012, 22, 17227–17235 RSC.
- T. Jin, F. Kitahara, S. Kamijo and Y. Yamamoto, Tetrahedron Lett., 2008, 49, 2824–2827 CrossRef CAS PubMed.
- S. M. Agawane and J. M. Nagarkar, Catal. Sci. Technol., 2012, 2, 1324–1327 CAS.
- O. Marvi, A. Alizadeh and S. Zarrabi, Bull. Korean Chem. Soc., 2011, 32, 4001–4004 CrossRef CAS.
- J. Lasri, M. J. F. Rodríguez, M. F. C. G. da Silva, P. Smolenski, M. N. Kopylovich, J. J. R. F. da Silva and A. J. L. Pombeiro, J. Organomet. Chem., 2011, 696, 3513–3520 CrossRef CAS PubMed.
- L. V. Myznikov, Y. A. Efimova, T. V. Artamonova and G. I. Koldobskii, Russ. J. Org. Chem., 2011, 47, 728 CrossRef CAS.
- P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525–3528 CrossRef CAS PubMed.
- S. Mukhopadhyay, J. Lasri, M. A. J. Charmier, M. F. C. G. da Silvaa and A. J. L. Pombeiro, Dalton Trans., 2007, 5297–5304 RSC.
- (a) F. Sen, Y. Karatas, M. Gulcan and M. Zahmakiran, RSC Adv., 2014, 4, 1526–1531 RSC; (b) İ. Esirden, E. Erken, M. Kaya and F. Sen, Catal. Sci. Technol., 2015 10.1039/c5cy00864f; (c) H. Pamuk, B. Aday, F. Şen and M. Kaya, RSC Adv., 2015, 5, 49295 RSC; (d) E. Erken, H. Pamuk, Ö. Karatepe, G. Başkaya, H. Sert, O. M. Kalfa and F. Şen, J. Cluster Sci., 2015 DOI:10.1007/s10876-015-0892-8; (e) F. Şen and G. Gökağaç, Energy Fuels, 2008, 22, 1858–1864 CrossRef; (f) Z. Ozturk, F. Sen, S. Sen and G. Gokagac, J. Mater. Sci., 2012, 47, 8134–8144 CrossRef CAS.
- V. Rama, K. Kanagaraj and K. Pitchumani, J. Org. Chem., 2011, 76, 9090–9095 CrossRef CAS PubMed.
- F. Sen, S. Sen and G. Gökağaç, Phys. Chem. Chem. Phys., 2011, 13, 1676–1684 RSC.
- F. Sen and G. Gökağaç, J. Appl. Electrochem., 2014, 44, 199–207 CrossRef CAS.
- S. Ertan, F. Sen, G. Gökağaç and S. Sen, J. Nanopart. Res., 2013, 15, 1979 CrossRef.
- S. Ertan, F. Sen, S. Sen and G. Gökağaç, J. Nanopart. Res., 2012, 14, 922 CrossRef.
- S. Sen, F. Sen and G. Gökağaç, Phys. Chem. Chem. Phys., 2011, 13, 6784–6792 RSC.
- F. Sen and G. Gökağaç, J. Phys. Chem. C, 2007, 111, 1467–1473 CAS.
- F. Sen and G. Gökağaç, J. Phys. Chem. C, 2007, 111, 5715–5720 CAS.
- A. Khalafi-Nezhad and S. Mohammadi, RSC Adv., 2013, 3, 4362–4371 RSC.
- B. Sreedhar, A. S. Kumar and D. Yada, Tetrahedron Lett., 2011, 52, 3565–3569 CrossRef CAS PubMed.
- L. Lang, H. Zhou, M. Xue, X. Wang and Z. Xu, Mater. Lett., 2013, 106, 443–446 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11426h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |