
Iodine catalyzed one-pot synthesis of highly substituted N-methyl pyrroles via [3 + 2] annulation and their in vitro evaluation as antibacterial agents†
Biguvu Balachandraa,
Sivakumar Shanmugam*a,
Thillaichidambaram Muneeswaranb and
Muthiah Ramakritinanb
aDepartment of Organic Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai – 625 021, Tamil Nadu, India. E-mail: shivazzen@mkuniversity.org; Tel: + 91 452 245 8471 ext. 371
bDepartment of Marine and Coastal Studies, School of Energy, Environment and Natural Resources, Madurai Kamaraj University, Madurai – 625 021, Tamil Nadu, India
First published on 15th July 2015
Abstract
A new class of highly substituted pyrroles have been synthesized via a simple, fast, and efficient method using environmentally friendly iodine catalyzed [3 + 2] annulation. N-Methyl-N-[(E)-1-(methylsulfanyl)-2-nitro-1-ethenyl]amine (NMSM) 1 and β-nitrostyrenes 3 underwent cycloaddition to afford the desired products 4 in excellent yields under solvent and metal free conditions. All the pyrrole derivatives were evaluated for their in vitro anti-bacterial activity. Among the synthesized pyrrole derivatives, 4b, 4c, 4e, 4g, 4i, 4j, 4l, 4m and 4n displayed good inhibitory properties against a panel of Gram positive and negative infectious pathogens.
Introduction
Over the past decades, the design and synthesis of substituted pyrroles has been important for obtaining building blocks for organic synthesis. This key heterocyclic core is found in a large number of natural and unnatural compounds, which has significant importance in pharmacology and materials science. Besides the natural products and their analogues, unnatural pyrroles show attractive biological activities, and are present in many bioactive compounds like HIV fusion inhibitors,1a antitubercular compounds,1b–c non-steroidal anti-inflammatory compound tolmetin and cholesterol-lowering agent atorvastatin, which is one of the top selling drugs worldwide (Fig. 1).1d,e Substituted pyrrole derivatives show more biological activities2 like antioxidant,3 anti-inflammatory,4 antibacterial,5 antifungal6 and antitumor7 activities etc. N-Methyl substituted heterocycles are significant synthetic targets owing to their wide range of applications as medicinal compounds and they can also modulate the physical and biological properties of the molecule. Methyl homologation increases the inhibitory potency of HMG-COAR inhibitors.8a–d S-Methyl substituted pyrrole derivatives and their analogues are useful precursors for synthesizing H2 receptor histamine antagonists.8e–g As a result, methods for the preparation of N- and S-methyl substituted heterocycle-containing structural scaffolds are in high demand.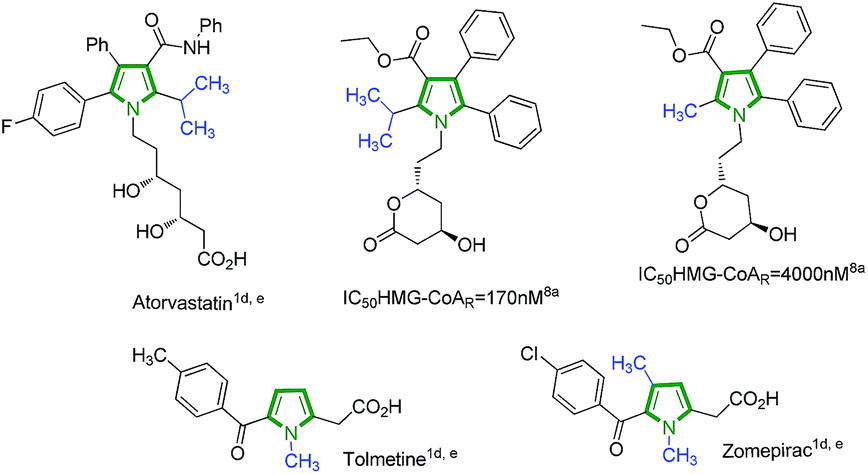 | ||
| Fig. 1 Pyrrole based biologically important compounds. | ||
In general, the standard methods to synthesise pyrrole are the Hantzsch,9 Knorr & Paal–Knorr10 methods, and multi-component synthesis,11 tandem reactions,21 transition-metal-catalyzed cyclization reactions13 etc. These methods allow the efficient construction of pyrroles with various substitution patterns, atom economy and regioselectivity. Despite the number of available synthetic strategies and their advantages, modern methodologies are focused on the solvent and metal free synthesis of substituted pyrroles due to lower energy consumption, increased selectivity, and minimized waste, hazards, toxicity and cost.14 Recently, iodine catalyzed reactions have been of considerable interest for various organic transformations due to its low toxicity to the environment, ready availability and inexpensiveness. Therefore, it is worth contributing to the creation of environmentally benign processes.15 In light of literature precedent,1–15 it would be interesting to develop a useful and efficient approach to the synthesis of N- and S-methyl substituted pyrrole derivatives, which have a major effect on the partitioning into biological membranes16a,b for example, methyl thio-ethers and their sulfoxides and sulfones are commonly occurring components in biologically active molecules.16c,d N-Methyl-N-[(E)-1-(methylsulfanyl)-2-nitro-1-ethenyl]amine (NMSM) 1 is used on an industrial scale for the manufacture of anti-ulcer (histamine H2 receptor antagonist) bulk drugs ranitidine17 and nizatidine.18a NMSM 1 (ref. 18b and c) is a multi-faceted building block in organic synthesis.
The methyl sulfanyl group is an electron donor as well as a good leaving group and it could be replaced with a variety of nucleophiles following a nucleophilic vinylic substitution (SNV) mechanism.19 In the current study, we report the synthesis of highly functionalized N-methyl substituted pyrrole derivatives 4 via one-pot [3 + 2] cycloaddition of NMSM 1 and β-nitrostyrenes 3 under solvent free conditions.
Results and discussion
To commence our study, the model reaction of NMSM 1, benzaldehyde 2a and nitromethane was performed in the absence of catalyst in a multi-component fashion. Eventually, we ended up with very little conversion to 4a and a 5% yield (Table 1, entry 1). Then, the reaction was carried out in the presence of solvents like DMSO and DMF, which was found to be counterproductive, under catalyst free conditions at reflux (Table 1, entry 2 & 3). For further optimization of the reaction conditions, the reaction was conducted in the presence of an iodine catalyst in DMF solvent and the results showed that the compounds obtained were in trace amounts (Table 1, entry 4). To improve the yield of 4a, different Lewis acids such as FeCl3, Yb(OTf)3 and CuI were used as catalysts to afford 4a in 15–25% yield (Table 1, entries 6–9). However, when molecular iodine was used as the catalyst (10 mol%), the target product 4a was obtained in 30% yield (Table 1 entry 5). Unfortunately, there was no significant improvement in the cycloaddition after increasing the temperature and amount of catalyst.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temp (°C) | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: 1 (1.0 mmol), 2a (1.0 mmol), nitromethane (1 ml) catalyst (10 mol%).b Isolated yields after column chromatography.c Starting material 1 was recovered. | |||||
| 1 | — | 90 | 9 | 5c | |
| 2 | — | 180 | DMSO | 12 | Traces |
| 3 | — | 145 | DMF | 12 | Traces |
| 4 | Iodine | 145 | DMF | 12 | Traces |
| 5 | Iodine | 90 | — | 9 | 30 |
| 6 | FeCl3 | 90 | — | 9 | 20c |
| 7 | Yb(OTf)3 | 90 | — | 9 | 25 |
| 8 | AcOH | 95 | — | 9 | 15c |
| 9 | CuI | 90 | — | 9 | 17c |
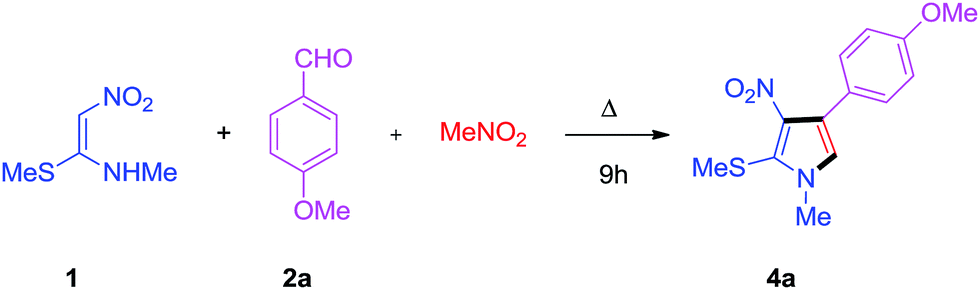
In order to improve the yield of 4a, the nitrostyrene20a was prepared separately and subjected to cycloaddition with NMSM 1 by manual grinding using mortar and pestle at room temperature. Initially, we tried a catalytic amount of anhydrous FeCl3 with 1 and 3a in a grinding method to afford 4a in 63% yield (Table 2, entry 1). We repeated the cycloaddition using metal catalysts such as AlCl3, CuI, ZnCl2, CuCl2·2H2O, and Yb(OTf)3 using the grinding method at RT, however, there was not much improvement in the yield of 4a (Table 2, entries 2–6).
|
|||
|---|---|---|---|
| Entry | Catalyst | Time (min) | Yieldb (%) |
| a Reaction conditions: 1 (1 mmol), 3a (1 mmol), catalyst (10 mol%).b Isolated yields after column chromatography.c Starting material 1 was recovered. | |||
| 1 | FeCl3 | 30 | 63 |
| 2 | AlCl3 | 30 | 52 |
| 3 | CuI | 30 | 50c |
| 4 | ZnCl2 | 30 | 53 |
| 5 | CuCl2·2H2O | 30 | 49c |
| 6 | Yb(OTf)3 | 30 | 65 |
| 7 | Iodine | 30 | 70 |
| 8 | Acetic acid | 30 | 65 |

Interestingly, the cycloaddition product 4a was obtained in 70% yield in the presence of an iodine catalyst (Table 2, entry 7). Using acetic acid, 4a was obtained in moderate yield (Table 2, entry 8).
We then tried an alternative method to study the tolerance of 3a and 1 in affording 4a, using different reaction conditions with various solvents and catalysts. The cycloaddition was conducted with NMSM 1 and 3a at 65 °C for 6 h under catalyst and solvent free conditions (Table 3, entry 1). However the yield was reduced to 25% on increasing the temperature from 65 °C to 90 °C (Table 3, entry 2). The yield of 4a was considerably increased to 54–62% yield, when the solvent was varied from water to ethanol under reflux conditions (Table 3, entries 4–8). When other Lewis acids were used as the catalyst under solvent free conditions, the desired product 4a was obtained in moderate yields (Table 3, entries 9–14). The compound 4a was obtained in 50% yield, when a catalytic amount of acetic acid was used (Table 3, entry 15). The solvent effect had not much influence on improving the product yield. Furthermore, to design an environmentally benign procedure, a model cycloaddition reaction was performed with 1 and 3a at 55 °C in the presence of iodine as the catalyst. Interestingly, the cycloaddition product 4a was obtained in 82% yield under solvent free conditions in 5 min (Table 3, entry 16). The yield of 4a decreased to 67% when reaction was carried out at 90 °C (Table 3 entry 17). Similarly, the yield of 4a decreased to 66% when the cycloaddition was performed in MeOH at reflux for 6 h (Table 3 entry 18) along with the iodine catalyst. Overall, iodine catalyzed cycloaddition (Tables 1–3) was found to be the better method for the synthesis of 4a.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1 (1 mmol), 3a (1 mmol), catalyst (10 mol%).b Isolated yields after column chromatography.c Starting material 1 was recovered. | |||||
| 1 | — | — | 65 | 6 | 32c |
| 2 | — | — | 90 | 5 | 25c |
| 3 | — | MeOH | RT | 48 | 10c |
| 4 | — | H2O | Reflux | 9 | 54 |
| 5 | — | MeOH | Reflux | 8 | 59 |
| 6 | — | EtOH | Reflux | 8 | 60 |
| 7 | FeCl3 | EtOH | Reflux | 8 | 60 |
| 8 | FeCl3 | MeOH | Reflux | 8 | 62 |
| 9 | FeCl3 | — | 50 | 30 min | 67 |
| 10 | AlCl3 | — | 55 | 30 min | 60 |
| 11 | CuI | — | 55 | 30 min | 61 |
| 12 | ZnCl2 | — | 55 | 30 min | 65 |
| 13 | CuCl2·2H2O | — | 55 | 30 min | 55 |
| 14 | Yb(OTf)3 | — | 55 | 45 | 72 |
| 15 | AcOH | — | 55 | 30 min | 50 |
| 16 | Iodine | — | 55 | 5 min | 82 |
| 17 | Iodine | — | 90 | 5 min | 67 |
| 18 | Iodine | MeOH | Reflux | 6 | 66 |

Among the above reactions, [3 + 2] cycloaddition under solvent free conditions at 55 °C was found to be the ideal method for the synthesis of tetra substituted pyrrole 4a in good yield (Table 3, entry 16). The NMR spectral data support the structure of 4a.
Following optimization of the reaction conditions, we investigated the scope of the reaction between NMSM 1 and a β-nitrostyrene 3 via [3 + 2] cycloaddition to afford 4a–n (Table 4). The starting materials containing electron-donating and -withdrawing groups on the β-nitrostyrene (3) were well tolerated under the optimized reaction conditions to afford 4a–n in moderate to good yields (65–90%). Temperature plays a significant effect in the reaction because the yields were improved to 65–90% at 55 °C (Table 4). Whether the aryl group of β-nitrostyrene 3 contains an electron-donating group or an electron-withdrawing group present at the ortho or meta positions, this shows little influence to afford 4c, 4f, 4h, 4j in similarly lower yields. Whereas, the products with the para substituents 4a, 4g, 4i, and 4k were obtained in high yield. An electron withdrawing group such as –NO2 was well tolerated under the optimized conditions and gave a moderate yield of 65% of the desired product 4m (Table 4). A halogen substituted phenyl group of the β-nitrostyrene 3 leads to the formation of pyrroles 4g and 4i in 83% yield. A naphthyl substituted β-nitrostyrene works well for the formation of pyrrole 4n and gave the highest yield 90% (Table 4). In conclusion all types of β-nitrostyrenes could be successfully applied in this reaction providing N-methylated pyrroles 4a–4n in good yield.
|
|---|
| a Reaction conditions: 1 (1 mmol), 3 (1 mmol), catalyst (10 mol%). Isolated yields after column chromatography. |
 |

Interestingly, nitro- and thioether linkage-containing structural scaffolds are able to undergo further coupling reactions.20b–f In our present report, the synthesis of tetra substituted pyrroles (4) is assumed to get more attention as they are important intermediates in organic, biological, as well as materials chemistry. All the synthesized compounds (4) were well characterized using their IR and NMR (1H, 13C, DEPT-135) spectra. The 1H NMR spectrum of 4a is explained as an example. The 1H NMR spectrum shows two doublets at δ 7.28 ppm (d, J = 9.0 Hz, 2H) and 6.91 ppm (d, J = 8.6 Hz, 2H) for the para methoxy substituted phenyl group and the aromatic proton of the substituted pyrrole (C5, H) appears as a singlet at 6.67 ppm. The N-methyl, S-methyl and methoxy protons appear as singlets at 3.83 ppm, 2.49 ppm and 3.79 ppm respectively. The singlet at δ = 6.67 ppm (C-5 proton) is the diagnostic signal for 4a.
On the basis of previous literature reports,21 a plausible mechanism was proposed for the iodine catalyzed cycloaddition of 1 and 3 to yield 4 (Scheme 1). Here, iodine acts as a mild Lewis acid, which increases the nucleophilicity of NMSM 1. In the first step, due to the polarized push–pull alkene of NMSM 1, it undergoes Michael addition with 3 to form a new C–C single bond to form B. Then, the lone pair of the sulphur atom of the methyl sulfanyl group shifts its electrons towards the nitrogen, which causes intramolecular nucleophilic attack of the nitrogen and formation of new N–C bond to give C. Intermediate C undergoes elimination of H2O to give intermediate D, which on further elimination of nitrosyl hydride (HNO) leads to the formation of product 4.
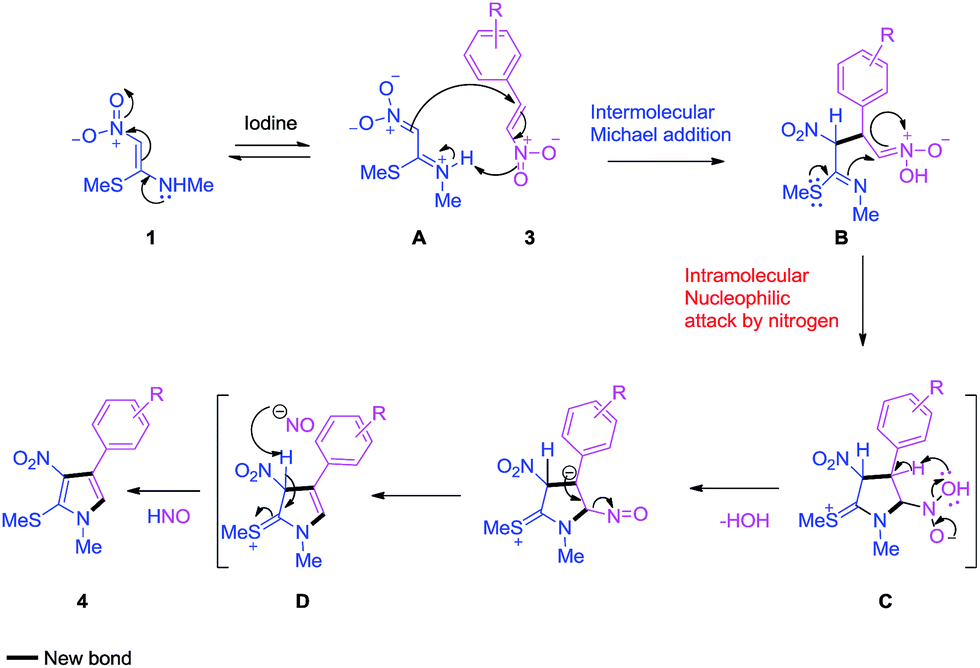 | ||
| Scheme 1 Plausible reaction mechanism for the formation of 4. | ||
Biological data
Disc diffusion method
Based on a biological literature survey, the synthesized compounds (4a–n) were evaluated for their anti-bacterial activity against selected Gram positive and negative bacteria, which are individually responsible for various infections and disorders, by using the standard disk diffusion assay22 described by Murray et al. in 1995. The Gram negative bacteria such as Salmonella typhi, Escherichia coli and Pseudomonas aeruginosa cause typhoid fever,23 haemolytic uremic syndrome,24 and nosocomial infections,25 respectively. The Gram positive bacteria Streptococcus pneumoniae is responsible for bronchitis, rhinitis, acute sinusitis, otitis media, conjunctivitis, meningitis, bacteraemia, sepsis, osteomyelitis, septic arthritis, endocarditis, peritonitis, pericarditis, cellulitis, and brain abscesses.26 Bacillus subtilis, a Gram positive model organism, causes food poisoning.27 Bacillus cereus causes food borne illnesses, causing severe nausea, vomiting, and diarrhoea and is responsible for “fried rice syndrome”.28 Stock solutions of the synthesized compounds were prepared in DMSO and filter sterilized using a 0.45 μm syringe filter. Briefly, cultures grown overnight containing 108 CFU ml−1 were spread on Mueller Hinton agar plates. Sterile paper discs (Himedia lab) were impregnated with the filter sterilized compounds, approximately 20 μL per disc. The paper discs were placed on the agar plates and incubated at 30 °C for 24 h. After 24 h of incubation, the zone of inhibition around the discs was observed and measured (Table 5). The values presented in the table are the average of two independent tests.| Entry | Compound code | Gram negativea | Gram positivea | ||||
|---|---|---|---|---|---|---|---|
| S. typhi | E. coli | P. aeruginosa | S. pneumonia | B. subtilis | B. cereus | ||
| a The values represent the activity of the compounds against the bacteria (with a 0.01 M solution), and show the zone of inhibition by diameter (mm). –: no inhibition.b Streptomycin | |||||||
| 1 | 4a | — | — | — | — | — | — |
| 2 | 4b | — | — | — | — | 6 | 9 |
| 3 | 4c | — | — | — | 9 | 9 | — |
| 4 | 4d | — | — | — | — | — | — |
| 5 | 4e | 7 | 8 | 7 | 9 | 9 | 8 |
| 6 | 4f | — | — | — | — | — | — |
| 7 | 4g | — | — | — | 8 | 11 | — |
| 8 | 4h | — | — | — | — | — | — |
| 9 | 4i | 7 | — | — | — | — | — |
| 10 | 4j | 8 | — | — | — | — | — |
| 11 | 4k | — | — | — | — | — | — |
| 12 | 4l | 6 | — | 9 | 11 | 8 | 8 |
| 13 | 4m | 9 | — | — | — | — | — |
| 14 | 4n | — | — | — | 7 | 7 | 8 |
| 15 | Cont. l | — | — | — | — | — | — |
| 16 | Stdb | 14 | 10 | 15 | 14 | 18 | 16 |
The results of the initial antibacterial activity screening revealed that among the pyrrole derivatives the compounds 4b, 4c, 4e, 4g, 4i, 4j, 4l, 4m and 4n displayed activity against the Gram-positive bacteria and Gram-negative bacteria, inhibitory zones (6–11 mm) are shown in Table 5. The compounds 4e and 4l showed good activity against almost all bacteria. Whereas the compound 4g displayed good activity against Gram-positive bacteria (Table 5, entry 7). These results provoked considerable interest to find the minimum inhibitory concentration (MIC).
Minimum inhibitory concentration (MIC)
The compounds which showed sensitivity to the bacteria were selected for the determination of the MIC (Fig. 2).29 The cultures grown overnight were adjusted to an OD of 0.1. Stock solutions of the synthesized compounds and standards were serially diluted to achieve a concentration between 10 mM to 0.001 mM and 1 to 10 μg ml−1 respectively. The MIC values of the compounds were determined by adopting the micro-well dilution method (Zgoda and Porter, 2001).30 Briefly, each well of the 96 well plates was seeded with the serially diluted compound, the bacterial culture and nutrient broth to a final volume of 200 μL per well. A well containing the cells and nutrient broth served as a negative control. Similarly a well seeded with DMSO, the cells and nutrient broth served as a solvent control. The plates were sealed tightly with a sterile plate sealer and incubated for 24 h in an orbital shaker at 90 rpm. Bacterial growth was measured using the optical density (OD) at 600 nm using 96 well plate readers (ELISA Plate reader) and also using the visual appearance of turbidity. Further confirmation was obtained by plating 10 μL of the samples from the clear wells on nutrient agar.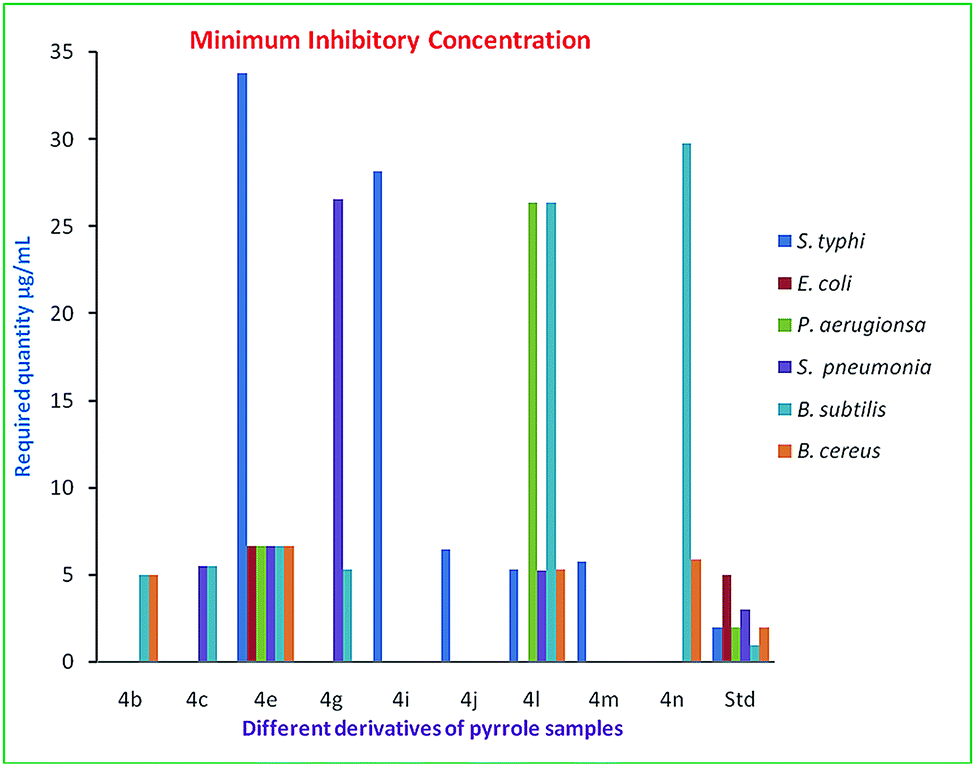 | ||
| Fig. 2 Minimum inhibitory concentration of the selected compounds. | ||
MIC values are defined as the lowest concentration that completely inhibits visible growth of a microorganism. Compound 4b displayed activity against Bacillus cereus and Bacillus subtilis (Gram-positive bacteria) due the presence of the phenyl substituent on the pyrrole ring (Table 6, entry 1). Moreover, the 3-methoxy substituted aryl group of pyrrole 4c showed considerable activity against Streptococcus pneumoniae and Bacillus subtilis (Table 6, entry 2). Whereas the 3,4,5-trimethoxy compound 4e displayed activity against both the Gram-positive bacteria and Gram-negative bacteria (Table 6, entry 3). The halogen substituted phenyl derivatives of 4 showed good antibacterial activities. Among the halogens, the 4-fluoro derivative (4g) was the most effective for Bacillus subtilis and has considerable activity against Streptococcus pneumoniae (Table 6, entry 4). Whereas 4i (4-chloro) and 4j (2-bromo) showed moderate activities for the Gram-negative bacteria Salmonella typhi (Table 6, entry 5 and 6).
| Entry | Compound code | Gram negativea | Gram positivea | ||||
|---|---|---|---|---|---|---|---|
| S. typhi | E. coli | P. aeruginosa | S. pneumonia | B. subtilis | B. cereus | ||
| a The values show considerable activity, and the quantity in μg ml−1 required to confine the bacterial growth. –: no inhibition.b Streptomycin | |||||||
| 1 | 4b | — | — | 5.0 | 5.0 | ||
| 2 | 4c | — | — | — | 5.5 | 5.5 | — |
| 3 | 4e | 33.8 | 6.7 | 6.7 | 6.7 | 6.7 | 6.7 |
| 4 | 4g | — | — | — | 26.6 | 5.3 | — |
| 5 | 4i | 28.2 | — | — | — | — | — |
| 6 | 4j | 6.5 | — | — | — | — | — |
| 7 | 4l | 5.3 | — | 26.4 | 5.3 | 26.4 | 5.3 |
| 8 | 4m | 5.8 | — | — | — | — | — |
| 9 | 4n | — | — | — | — | 29.8 | 5.9 |
| 10 | Stdb | 2.0 | 5.0 | 2.0 | 3.0 | 1.0 | 2.0 |
Interestingly the 3-hydroxy substituted aryl group of 4l showed a wide range of activity (Table 6, entry 7), but the 4-nitro substituted aryl group of 4m was weakly effective against Salmonella typhi (Table 6, entry 8). The compound 4n with naphthyl substitution displayed activity against Bacillus cereus and Bacillus subtilis (Gram-positive bacteria) (Table 6, entry 9). Due to the strong resistance of the pathogens towards the anti-bacterial agent, some of the pyrrole derivatives did not show inhibitory properties against Gram negative and positive bacteria (Tables 5 and 6). The obtained results show that different substituents influence the activity of the N-methyl substituted pyrrole compounds.
Conclusion
Hence we have developed a simple, fast, and efficient method for the synthesis of tetra substituted pyrroles in the presence of a catalytic amount of iodine under metal and solvent free conditions. In comparison with reported procedures, the present one affords an environmentally benign approach for the synthesis of pyrrole derivatives 4a–n. In this procedure new C–C and C–N bonds were effectively constructed. The presence of nitro and sulphur groups enables further construction of complex derivatives. The synthesized compounds 4a–n were evaluated for their anti-bacterial activity against selected bacteria. Most of the synthesized compounds have good antibacterial activity.Experimental section
General considerations
Melting points were determined using open capillary tubes and were uncorrected. IR spectra were obtained on a Jasco FT-IR instrument using KBr pellets and the data are reported in cm−1. Mass spectra were obtained with an Agilent mass spectrometer and recorded in positive and negative mode with an ESI source. The 1H and 13C NMR spectra of the new compounds were measured at 300 MHz and 75 MHz in CDCl3 and DMSO-d6 with TMS as the internal standard. The chemical shifts are expressed in ppm, the coupling constants (J values) are given in Hertz (Hz) and the spin multiplicities are indicated by the following symbols: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), td (triplet of doublets). Elemental analyses were carried out with a Perkin Elmer 2400 Series II analyzer. Silica gel-G plates (Merck) were used for TLC analysis with a mixture of petroleum ether (60–80 °C) and ethyl acetate as the eluent. All chemicals were purchased and used without further purification. The starting material β-nitrostyrenes 3 were prepared according to the previous literature methods. Nitroketene-N,S-acetal (NMSM) 1 is commercially available and was used without further purification.General procedure for the preparation of the β-nitrostyrenes (3)
Aldehyde (0.05 mmol), nitromethane (0.05 mmol), and MeOH (10–20 ml) were added to a round-bottom flask and then stirred vigorously. NaOH solution (10.5 M, 10 ml) was added dropwise to the mixture in an ice bath; a large amount of yellow solid precipitated, and the stirring was continued for 15 min. Distilled H2O was added until the solution became clear, and then the solution was added dropwise to concentrated HCl (30 ml), and a yellow solid precipitated. The yellow solid was filtered and washed with H2O, then the solvent was evaporated in a vacuum drying oven. After recrystallization (EtOH), yellow needle-like crystals were obtained. 4-OMe-β-nitrostyrene (3a): isolated yield of 93%; yellow solid; mp 86–88 °C; 1H NMR (300 MHz, CDCl3): δ, 7.98 (d, J = 13.6 Hz, 1H), 7.52 (m, 3H), 6.96 (d, J = 8.8 Hz, 2H), 3.87 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 162.88, 138.97, 134.91, 131.11, 122.44, 114.84, and 55.45. The other β-nitrostyrene derivatives were prepared using a similar procedure and characterized by 1H and 13C NMR.General procedure for the synthesis of the substituted pyrroles (4)
NMSM 1 (1.0 mmol), β-nitrostyrene 3 (1.0 mmol) and molecular iodine (10 mol%) were charged into a 25 ml glass vial equipped with a stirring bar. The reaction mixture was heated in an oil bath at 55 °C for 5–10 min (monitored by TLC). After cooling down to room temperature, the resulting mixture was extracted with ethyl acetate (3 × 7 ml), and then washed with water (2 × 10 ml) followed by brine solution (1 × 15 ml). The organic phases were collected and dried with anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified using column chromatography on silica gel (petroleum ether/EtOAc) to afford the corresponding product 4.Experimental procedure for the three component one pot synthesis of the substituted pyrrole (Table 1)
Aldehyde (1.0 mmol), nitromethane (1 ml), and molecular iodine (10 mol%) were charged into a 25 ml glass vial equipped with a stirring bar. The reaction mixture was allowed to reflux at 90 °C for 50–60 min, then after cooling down to room temperature, NMSM 1 (1.0 mmol) was added by sequential addition and the refluxing was continued for 9 h (monitored by TLC). The resulting mixture was extracted with ethyl acetate (3 × 7 ml), and then washed with water (2 × 10 ml) followed by brine solution (1 × 15 ml). The organic phases were collected and dried with anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified using column chromatography on silica gel (petroleum ether/EtOAc) to afford the corresponding product 4a.Experimental procedure for the one pot synthesis of the substituted pyrrole using a manual grinding method (Table 2)
NMSM 1 (1.0 mmol), β-nitrostyrene 3a (1.0 mmol) and the catalyst (10 mol%) were subjected to manual grinding for 30 min (monitored by TLC) at RT using a mortar and pestle. The resulting mixture was extracted with ethyl acetate (3 × 7 ml), and then washed with water (2 × 10 ml) followed by brine solution (1 × 15 ml). The organic phases were collected and dried with anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified using column chromatography on silica gel (petroleum ether/EtOAc) to afford the corresponding product 4a.4-(4-Methoxyphenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4a)
Yellow solid; mp 128–130 °C; yield: 0.153 g (82%); 1H NMR (300 MHz, CDCl3): δ, 7.28 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 8.6 Hz, 2H), 6.67 (s, 1H), 3.83 (s, 3H), 3.79 (s, 3H), 2.49 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 158.88, 136.99, 129.74, 126.09, 124.47, 122.17, 121.76, 113.55, 55.14, 34.87, 19.38; IR (ATR KBr cell, cm−1): 791, 1327, 1495, 3741, 3840; anal. calcd for C13H14N2O3S: C, 56.10; H, 5.07; N, 10.06; found: C, 56.01; H, 5.03; N, 10.02; LC-MS (ESI) calcd m/z: 278, found 279 [(M + H)]+.1-Methyl-2-(methylthio)-3-nitro-4-phenyl-1H-pyrrole (4b)
Yellow solid; mp 129–131 °C; yield: 0.120 g (72%); 1H NMR (300 MHz, CDCl3): δ, 7.35 (s, 5H), 6.71 (s, 1H), 3.80 (s, 3H), 2.50 (s, 3H). 13C NMR (75 MHz, CDCl3): δ, 137.00, 132.06, 128.46, 128.06, 127.20, 126.30, 122.32, 122.06, 34.88, 19.38; IR (ATR KBr cell, cm−1): 691, 756, 1322, 1479, 2348, 3728. Anal. calcd for C12H12N2O2S: C, 58.05; H, 4.87; N, 11.28; found: C, 58.03; H, 4.86; N, 11.26; LC-MS (ESI) calcd. m/z: 248, found 249 [(M + H)]+.4-(3-Methoxyphenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4c)
Yellow solid; mp 127–129 °C; yield: 0.150 g (80%); 1H NMR (300 MHz, CDCl3): δ, 7.27 (s, 1H), 6.91 (m 3H), 6.72 (s, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 2.50 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 159.38, 137.30, 133.46, 129.11, 126.24, 122.41, 121.96, 121.00, 114.25, 113.01, 55.18, 34.89, 19.46; IR (ATR KBr cell, cm−1): 688, 784, 1321, 1477, 2349, 2930; anal. calcd for C13H14N2O3S: C, 56.10; H, 5.07; N, 10.06; found: C, 56.01; H, 5.03; N, 10.02; LC-MS (ESI) calcd. m/z: 278, found 279 [(M + H)]+.4-(3,4-Dimethoxyphenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4d)
Yellow solid; mp 118–120 °C; yield: 0.174 g (84%); 1H NMR (300 MHz, CDCl3): δ, 6.93–6.85 (m, 3H), 6.70 (s, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.80 (s, 3H), 2.50 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 148.63, 137.22, 126.10, 124.91, 122.23, 121.97, 121.02, 112.53, 111.13, 110.89, 55.90, 55.86, 34.85, 19.42; IR (ATR KBr cell, cm−1): 804, 1240, 1504, 3741, 3840; anal. calcd for C14H16N2O4S: C, 54.53; H, 5.23; N, 9.08; found: C, 54.50; H, 5.21; N, 9.05; LC-MS (ESI) calcd. m/z: 308, found 309 [(M + H)]+.1-Methyl-2-(methylthio)-3-nitro-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole (4e)
Yellow solid; mp 132–134 °C; yield: 0.198 g (87%); 1H NMR (300 MHz, CDCl3): δ, 6.73 (s, 1H), 6.57 (s, 2H), 3.88 (s, 3H), 3.86 (s, 6H), 3.80 (s, 3H), 2.51 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 152.85, 137.43, 137.04, 127.68, 126.42, 122.44, 122.06, 105.96, 60.77, 56.04, 34.93, 19.44; IR (ATR KBr cell, cm−1): 698, 820, 1105, 1339, 1497, 3741, 3840; anal. calcd for C15H18N2O5S: C, 53.24; H, 5.36; N, 8.28; found: C, 53.22; H, 5.34; N, 8.25; LC-MS (ESI) calcd. m/z: 338, found 339 [(M + H)]+.4-(2-Fluorophenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4f)
Yellow solid; mp 218–220 °C; yield: 0.138 g (77%); 1H NMR (300 MHz, CDCl3): δ, 7.38–7.24 (m, 2H), 7.20–7.04 (m, 2H), 6.76 (s, 1H), 3.81 (s, 3H), 2.51 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 161.60, 158.32, 130.60, 129.37, 129.26, 126.49, 123.97, 122.98, 120.49, 120.29, 115.64, 115.34, 35.12, 19.48; IR (ATR KBr cell, cm−1): 636, 767, 1329, 1480, 2348, 3728; anal. calcd for C12H11FN2O2S: C, 54.12; H, 4.16; N, 10.52; found: C, 54.11; H, 4.14; N, 10.51; LC-MS (ESI) calcd. m/z: 266, found 267 [(M + H)]+.4-(4-Fluorophenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4g)
Yellow solid; mp 168–170 °C; yield: 0.149 g (83%); 1H NMR (300 MHz, CDCl3): δ, 7.32 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 6.69 (s, 1H), 3.80 (s, 3H), 2.50 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 163.84, 160.58, 130.45, 130.34, 128.24, 126.70, 122.42, 121.27, 115.23, 114.94, 35.03, 19.42; IR (ATR KBr cell, cm−1): 793, 829, 1210, 1322, 1489, 3741, 3840; anal. calcd for C12H11FN2O2S: C, 54.12; H, 4.16; N, 10.52; found: C, 54.10; H, 4.13; N, 10.50; LC-MS (ESI) calcd. m/z: 266, found 267 [(M + H)]+.4-(3-Chlorophenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4h)
Yellow solid; mp 96–98 °C; yield: 0.142 g (75%); 1H NMR (300 MHz, CDCl3): δ, 7.33 (s, 1H), 7.27 (m, 3H), 6.72 (s, 1H), 3.80, (s, 3H), 2.50 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 136.64, 133.98, 133.62, 129.21, 128.41, 127.11, 126.94, 126.79, 122.74, 120.47, 34.93, 19.26; IR (ATR KBr cell, cm−1): 778, 1312, 1478, 3741, 3840; anal. calcd for C12H11ClN2O2S: C, 50.97; H, 3.92; N, 9.91; found: C, 50.95; H, 3.91; N, 9.90; LC-MS (ESI) calcd. m/z: 282, found 283 [(M + H)]+.4-(4-Chlorophenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4i)
Yellow solid; mp 76–78 °C; yield: 0.158 g (83%); 1H NMR (300 MHz, CDCl3): δ, 7.34 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 7.1 Hz, 2H), 6.70 (s, 1H), 3.80 (s, 3H), 2.50 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 136.89, 133.25, 130.72, 129.95, 128.30, 126.94, 122.48, 121.02, 35.05, 19.42; IR (ATR KBr cell, cm−1): 832, 1320, 1487, 3741, 3840; anal. calcd for C12H11ClN2O2S: C, 50.97; H, 3.92; N, 9.91; found: C, 50.95; H, 3.91; N, 9.90; LC-MS (ESI) calcd. m/z: 282, found 283 [(M + H)]+.4-(2-Bromophenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4j)
Yellow solid; mp 188–190 °C; yield: 0.170 g (77%); 1H NMR (300 MHz, CDCl3): δ, 7.63 (d, J = 7.9 Hz, 1H), 7.39–7.13 (m, 3H), 6.68 (s, 1H), 3.82 (s, 3H), 2.52 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 134.01, 132.46, 131.24, 129.08, 127.05*, 126.45, 124.60, 122.87, 121.15, 35.15, 19.34; IR (ATR KBr cell, cm−1): 764, 1325, 1479, 2347, 3615, 3728; anal. calcd for C12H11BrN2O2S: C, 44.05; H, 3.39; N, 8.56; found: C, 44.03; H, 3.36; N, 8.55; LC-MS (ESI) calcd. m/z: 327, found 328 [(M + H)]+ [* – two carbon signals have merged together].4-(4-Bromophenyl)-1-methyl-2-(methylthio)-3-nitro-1H-pyrrole (4k)
Yellow solid; mp 102–104 °C; yield: 0.174 g (79%); 1H NMR (300 MHz, CDCl3): δ, 7.49 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 8.5 Hz, 2H), 6.70 (s, 1H), 3.80 (s, 3H), 2.50 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 136.83, 131.23, 131.19, 130.25, 126.99, 122.45, 121.40, 121.00, 35.07, 19.42; IR (ATR KBr cell, cm−1): 617, 781, 1320, 1439, 3741, 3839; anal. calcd for C12H11BrN2O2S: C, 44.05; H, 3.39; N, 8.56; found: C, 44.02; H, 3.37; N, 8.54; LC-MS (ESI) calcd. m/z: 327, found 328 [(M + H)]+.3-(1-Methyl-5-(methylthio)-4-nitro-1H-pyrrol-3-yl)phenol (4l)
Yellow solid; mp 140–142 °C; yield: 0.151 g (85%); 1H NMR (300 MHz, CDCl3): δ, 7.33–7.15 (m, 1H), 6.90 (d, J = 7.4 Hz, 1H), 6.81 (d, J = 11.8 Hz, 1H), 6.70 (s, 1H), 5.01 (s, 1H), 3.79 (s, 3H), 2.49 (s, 3H); 13C NMR (75 MHz, DMSO): δ, 157.38, 136.71, 133.46, 129.44, 125.70, 123.91, 120.59, 118.98, 115.17, 114.30, 35.11, 19.33; IR (ATR KBr cell, cm−1): 774, 1306, 1472, 3447, 3741, 3840; anal. calcd for C12H12N2O3S: C, 54.53; H, 4.58; N, 10.60; found: C, 54.51; H, 4.56; N, 10.58; LC-MS (ESI) calcd. m/z: 264, found 265 [(M + H)]+.1-Methyl-2-(methylthio)-3-nitro-4-(4-nitrophenyl)-1H-pyrrole (4m)
Yellow solid; mp 258–260 °C; yield: 0.128 g (65%); 1H NMR (300 MHz, CDCl3): δ, 8.23 (d, J = 8.9 Hz, 2H), 7.50 (d, J = 8.9 Hz, 2H), 6.82 (s, 1H), 3.84 (s, 3H), 2.53 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 146.83, 139.15, 136.85, 129.31, 128.23, 123.45, 123.09, 120.09, 35.29, 19.44; IR (ATR KBr cell, cm−1): 807, 1321, 1493, 2348, 3729; anal. calcd for C12H11N3O4S: C, 49.14; H, 3.78; N, 14.33; found: C, 49.13; H, 3.76; N, 14.34; LC-MS (ESI) calcd. m/z: 293, found 294 [(M + H)]+.1-Methyl-2-(methylthio)-4-(naphthalen-1-yl)-3-nitro-1H-pyrrole (4n)
Orange solid; mp 168–170 °C; yield: 0.181 g (90%); 1H NMR (300 MHz, CDCl3): δ, 7.91–7.82 (m, 2H), 7.68 (d, J = 8.2 Hz, 1H), 7.52–7.34 (m, 4H), 6.74 (s, 1H), 3.86 (s, 3H), 2.56 (s, 3H); 13C NMR (75 MHz, CDCl3): δ, 138.20, 133.35, 132.45, 130.58, 128.25, 128.15, 127.31, 126.28, 126.12, 125.71, 125.29, 125.07, 123.46, 120.24, 35.05, 19.31; IR (ATR KBr cell, cm−1): 780, 1324, 1481, 3741, 3840; anal. calcd for C16H14N2O2S: C, 64.41; H, 4.73; N, 9.39; found: C, 64.40; H, 4.71; N, 9.37; LC-MS (ESI) calcd. m/z: 298, found 299 [(M + H)]+.Acknowledgements
SS thanks DST and UGC-MRP, New Delhi, for financial assistance. We thank DST-IRHPA for funding towards a high resolution NMR spectrometer. BBC thanks the University Grants Commission, New Delhi, for the award of Senior Research Fellowship.Notes and references
- (a) C. Teixeira, F. Barbault, J. Rebehmed, K. Liu, L. Xie, H. Lu, S. Jiang, B. Fan and F. Maurel, Bioorg. Med. Chem., 2008, 16, 3039–3048 CrossRef CAS PubMed; (b) M. Biava, G. C. Porretta, D. Deidda, R. Pompei, A. Tafic and F. Manettic, Bioorg. Med. Chem., 2004, 12, 1453–1458 CrossRef CAS PubMed; (c) M. Protopopova, E. Bogatcheva, B. Nikonenko, S. Hundert, L. Einck and C. A. Nacy, Med. Chem., 2007, 3, 301–316 CrossRef CAS; (d) V. Este’vez, M. Villacampa and J. C. Mene’ndez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC; (e) V. Este’vez, M. Villacampa and J. C. Mene’ndez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC.
- (a) J. A. H. Lainton, J. W. Huffman, B. R. Martin and D. R. Compton, Tetrahedron Lett., 1995, 36, 1401–1404 CrossRef CAS; (b) C. Y. DeLeon and B. Ganem, Tetrahedron, 1997, 53, 7731–7752 CrossRef CAS; (c) R. Martín, M. Rodríguez Rivero and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 7079–7082 CrossRef PubMed; (d) H. S. P. Rao and S. Sivakumar, Beilstein J. Org. Chem., 2007, 3, 31 CrossRef PubMed; (e) H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264–287 CrossRef CAS PubMed; (f) J. Deblander, S. V. Aeken, J. Jacobs, N. D. Kimpe and K. A. Tehrani, Eur. J. Org. Chem., 2009, 4882–4892 CrossRef CAS PubMed; (g) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585–9587 CrossRef CAS PubMed; (h) R. J. Billedeau, K. R. Klein, D. Kaplan and Y. Lou, Org. Lett., 2013, 15, 1421–1423 CrossRef CAS PubMed; (i) P. Dhanalakshmi and S. Sivakumar, RSC Adv., 2014, 4, 29493–29501 RSC; (j) M. Arun Divakar, V. Sudhamani, S. Sivakumar, T. Muneeswaran, S. Tamilzhalagan, M. Ramakritinan and K. Ganesan, RSC Adv., 2015, 5, 8362–8370 RSC.
- (a) A. S. Demir, I. M. Akhmedov and O. Sesenoglu, Tetrahedron, 2002, 58, 9793–9799 CrossRef CAS; (b) D. Wang, X. Hu and G. Zhao, Int. J. Food Sci. Technol., 2008, 43, 1880–1886 CrossRef CAS PubMed.
- (a) V. J. Demopoulos and E. J. Rekka, J. Pharmaceut. Sci., 1995, 84, 79–82 CrossRef CAS PubMed; (b) J. M. Muchowski, Adv. Med. Chem., 1992, 1, 109–135 CAS; (c) B. Khalili, P. Jajarmi, B. Eftekhari-Sis and M. M. Hashemi, J. Org. Chem., 2008, 73, 2090–2095 CrossRef CAS PubMed.
- R. W. Bürli, D. McMinn, J. A. Kaizerman, W. Hu, Y. Ge, Q. Pack, V. Jiang, M. Gross, M. Garcia, R. Tanaka and H. E. Moser, Bioorg. Med. Chem. Lett., 2004, 14, 1253–1257 CrossRef PubMed.
- (a) H. M. Meshram, B. R. V. Prasad and D. Aravind Kumar, Tetrahedron Lett., 2010, 51, 3477–3480 CrossRef CAS PubMed; (b) M. Z. Wang, H. Xu, T. W. Liu, Q. Feng, S. J. Yu, S. H. Wang and Z. M. Li, Eur. J. Med. Chem., 2011, 46, 1463–1472 CrossRef CAS PubMed; (c) A. K. Verma, S. K. Reddy Kotla, T. Aggarwal, S. Kumar, H. Nimesh and R. K. Tiwari, J. Org. Chem., 2013, 78, 5372–5384 CrossRef CAS PubMed.
- (a) E. Gimenez-Arnau, S. Missailidis and M. F. G. Stevens, Anti-Cancer Drug Des., 1998, 13, 431–451 CAS; (b) P. Cozzi and N. Mongelli, Curr. Pharm. Des., 1998, 4, 181–201 CAS; (c) Y. R. Torres, T. S. Bugni, R. G. S. Berlinck, C. M. Ireland, A. Magalhaes, A. G. Ferreira and R. M. Rocha, J. Org. Chem., 2002, 67, 5429–5432 CrossRef CAS PubMed; (d) B. Das, N. Bhunia and M. Lingaiah, Synthesis, 2011, 3471–3474 CrossRef CAS.
- (a) E. J. Barreiro, A. E. Kummerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; (b) H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed; (c) H. Schönherr and T. Cernak, Angew. Chem., 2013, 125, 12480–12492 CrossRef PubMed; (d) Z. J. Fu, Z. J. Li, Q. Xiong and H. Cai, Eur. J. Org. Chem., 2014, 7798–7802 CrossRef CAS PubMed; (e) M. L. Roantree and Y. R. Christopher, Eur. Pat. Appl., 5985, 12 Dec 1979; (f) R. C. Young, G. J. Durant, J. C. Emmett, C. R. Ganellin, M. J. Graham, R. C. Mitchell, H. D. Prain and M. L. Roantree, J. Med. Chem., 1986, 29, 44–49 CrossRef CAS; (g) R. C. Young, R. C. Mitchell, T. H. Brown, C. R. Ganellin, R. Griffiths, M. Jones, K. K. Rana, D. Saunders, I. R. Smith, N. E. Sore and T. J. Wilks, J. Med. Chem., 1988, 31, 656–671 CrossRef CAS.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474–1476 CrossRef PubMed.
- (a) L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635–1642 CrossRef PubMed; (b) C. Pall, Ber. Dtsch. Chem. Ges., 1885, 18, 367–371 CrossRef PubMed.
- (a) F. Benedetti, F. Berti, P. Nitti, G. Pitacco and E. Valentin, Gazz. Chim. Ital., 1990, 120, 25–28 CAS; (b) H. Shiraishi, T. Nishitani, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1998, 63, 6234–6238 CrossRef CAS PubMed; (c) G. Dou, C. Shi and D. Shi, J. Comb. Chem., 2008, 10, 810–813 CrossRef CAS PubMed; (d) Y. Lu and B. A. Arndtsen, Angew. Chem., Int. Ed., 2008, 47, 5430–5433 CrossRef CAS PubMed; (e) Y. J. Bian, X. Y. Liu, K. G. Ji, X. Z. Shu, L. N. Guo and Y. M. Liang, Tetrahedron, 2009, 65, 1424–1429 CrossRef CAS PubMed; (f) A. Aponick, C. Y. Li, J. Malinge and E. F. Marques, Org. Lett., 2009, 11, 4624–4627 CrossRef CAS PubMed; (g) S. L. A. Langle, M. Abarbri, J. Thibonnet, A. Duchene and J. L. Parrain, Chem. Commun., 2010, 46, 5157–5159 RSC; (h) W. Liu, H. Jiang and L. Huang, Org. Lett., 2010, 12, 312–315 CAS.
- (a) X. T. Liu, L. Huang, F. J. Zheng and Z. P. Zhana, Adv. Synth. Catal., 2008, 350, 2778–2788 CrossRef CAS PubMed; (b) Q. Cai, F. Zhou, T. Xu, L. Fu and K. Ding, Org. Lett., 2011, 13, 340–343 CrossRef CAS PubMed; (c) H. Ueda, M. Kameya, H. Yamaguchi, K. Sugimoto and H. Tokuyama, Org. Lett., 2014, 16, 4948–4951 CrossRef CAS PubMed.
- (a) M. Sukhendu, B. Srijit and J. Umasish, J. Org. Chem., 2010, 75, 1674–1683 CrossRef PubMed; (b) A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2010, 12, 372–374 CrossRef CAS PubMed; (c) A. Takahashi, S. Kawai, I. Hachiya and M. Shimizu, Eur. J. Org. Chem., 2010, 191–200 CrossRef CAS PubMed; (d) M. N. Zhao, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Org. Lett., 2014, 16, 608–611 CrossRef CAS PubMed.
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed; (b) M. S. Singh and S. Chowdhury, RSC Adv., 2012, 2, 4547–4592 RSC; (c) I. B. Subrahmanya and D. R. Trivedi, Tetrahedron Lett., 2013, 54, 5577–5582 CrossRef PubMed.
- (a) S. Tang, Y. Wu, W. Q. Liao, R. P. Bai, C. Liu and A. W. Lei, Chem. Commun., 2014, 50, 4496–4499 RSC; (b) A. Sagar, S. Vidyacharan and D. S. Sharada, RSC Adv., 2014, 4, 37047–37050 RSC; (c) S. Ko, M. N. V. Sastry, C. Lin and C. F. Yao, Tetrahedron Lett., 2005, 46, 5771–5774 CrossRef CAS PubMed; (d) C. M. Chu, S. Gao, M. N. V. Sastry and C. F. Yao, Tetrahedron Lett., 2005, 46, 4971–4974 CrossRef CAS PubMed; (e) B. K. Banik, M. Fernandez and C. Alvarez, Tetrahedron Lett., 2005, 46, 2479–2482 CrossRef CAS PubMed; (f) B. P. Bandgar and K. A. Shaikh, Tetrahedron Lett., 2003, 44, 1959–1961 CrossRef CAS; (g) J. S. Yadav, B. V. Subba Reddy, G. Narasimhulu, N. S. Reddy and P. Janardhan Reddy, Tetrahedron Lett., 2009, 50, 3760–3762 CrossRef CAS PubMed.
- (a) N. G. Anthony, K. R. Fox, B. F. Johnston, A. I. Khalaf, S. P. Mackay, I. S. McGroarty, J. A. Parkinson, G. G. Skellern, C. J. Suckling and R. D. Waigh, Bioorg. Med. Chem. Lett., 2004, 141, 1353–1356 CrossRef PubMed; (b) B. R. Beno, K. S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed; (c) C. S. Richards-Taylor, D. C. Blakemoreb and M. C. Willis, Chem. Sci., 2014, 5, 222–228 RSC; (d) B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shavnya, Org. Lett., 2014, 16, 154–157 CrossRef CAS PubMed.
- (a) M. J. Daly and B. J. Price, in Progress in Medicinal Chemistry, ed. G. P. Ellis and G. B. West, Elsevier, Amsterdam, 1983, vol. 20, pp. 337–368 Search PubMed; (b) Y. Ootsuka, H. Morita and H. Mori, Jpn. Kokai Tokyo Koho JP 07157465 A2, 1995, Chem. Abstr., 1995, 123, 767921.
- (a) K. P. Moder, Eur. Pat. Appl. EP 515121 A1, 1992, Chem. Abstr., 1993, 118, 169100; (b) P. Gunasekaran, P. Prasanna, S. Perumal and A. I. Almansour, Tetrahedron Lett., 2013, 54, 3248–3252 CrossRef CAS PubMed; (c) P. Gunasekaran, P. Prasanna, S. Perumal and A. I. Almansour, Tetrahedron Lett., 2014, 55, 329–332 CrossRef CAS PubMed.
- (a) C. Venkatesh, B. Singh, P. K. Mahata, H. Ila and H. Junjappa, Org. Lett., 2005, 7, 2169–2172 CrossRef CAS PubMed; (b) H. S. P. Rao and S. Sivakumar, J. Org. Chem., 2005, 70, 4524–4527 CrossRef CAS PubMed; (c) H. S. P. Rao and K. Geetha, Tetrahedron Lett., 2009, 50, 3836–3839 CrossRef CAS PubMed; (d) H. S. P. Rao, K. Geetha and M. Kamalraj, RSC Adv., 2011, 1, 1050–1059 RSC; (e) H. S. P. Rao and A. Parthiban, Org. Biomol. Chem., 2014, 12, 6223–6238 RSC; (f) H. S. P. Rao and A. V. B. Rao, Eur. J. Org. Chem., 2014, 3646–3655 CrossRef CAS PubMed.
- (a) G. Yan, A. J. Borah and L. Wang, Org. Biomol. Chem., 2014, 12, 6049–6058 RSC; (b) T. Sugahara, K. Murakami, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 9329–9333 CrossRef CAS PubMed; (c) L. Wang, W. Hea and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC; (d) Z. Fang, J. Liu, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2014, 53, 7209–7213 CrossRef CAS PubMed; (e) F. Zhu and Z.-X. Wang, Org. Lett., 2015, 17, 1601–1604 CrossRef CAS PubMed; (f) W. Zhang, J. Xie, B. Rao and M. Luo, J. Org. Chem., 2015, 80, 3504–3511 CrossRef CAS PubMed.
- (a) G. P. Desiraju and R. L. Harlow, J. Am. Chem. Soc., 1998, 111, 6757 CrossRef; (b) A. C. Legon, Angew. Chem., 1999, 38, 2686–2714 CrossRef; (c) M. Rueping and A. Parra, Org. Lett., 2010, 12, 5281–5283 CrossRef CAS PubMed; (d) G. R. Reddy, T. R. Reddy, S. C. Joseph, K. S. Reddy and M. Pal, RSC Adv., 2012, 2, 3387–3395 RSC; (e) P. Xu, K. Huang, Z. Liu, M. Zhou and W. Zeng, Tetrahedron Lett., 2013, 54, 2929–2933 CrossRef CAS PubMed; (f) A. Srivastava, G. Shukla, A. Nagaraju, G. K. Verma, K. Raghuvanshi, R. C. F. Jones and M. S. Singh, Org. Biomol. Chem., 2014, 12, 5484–5491 RSC.
- P. R. Murray, E. J. Baron, M. A. Pfaller, F. C. Tenover and R. H. Yolke, Manual of Clinical Microbiology, ASM, Washington DC, 6th edn, 1995 Search PubMed.
- (a) A. Kothari, A. Pruthi and T. D. Chugh, J. Infect. Dev. Countries, 2008, 4, 253–259 Search PubMed; (b) J. A. Crump, S. P. Luby and E. D. Mintz, Bull. W. H. O., 2004, 82, 346–353 Search PubMed.
- H. M. Verweyen, H. Karch, F. Allerberger and L. B. Zimmerhackl, Enterohemorrhagic Escherichia coli (EHEC) in pediatric hemolytic uremic syndrome: a prospective study in Germany and Austria, Infection, 1999, 27, 341–347 CrossRef CAS.
- (a) S. Bentzmann and P. Plésiat, Environ. Microbiol., 2011, 13, 1655–1665 CrossRef PubMed; (b) N. Cramer, J. Klockgether, K. Wrasman, M. Schmidt, C. F. Davenport and B. Tümmler, Environ. Microbiol., 2011, 13, 1690–1704 CrossRef CAS PubMed.
- R. A. C. Siemieniuk, D. B. Gregson and M. J. Gill, The persisting burden of invasive pneumococcal disease in HIV patients: an observational cohort study, BMC Infect. Dis., 2011, 11, 314, DOI:10.1186/1471-2334-11-314.
- N. A. Logan, Bacillus species of medical and veterinary importance, J. Med. Microbiol., 1988, 25, 157–165 CrossRef CAS.
- A. Kotiranta, K. Lounatmaa and M. Haapasalo, Epidemiology and pathogenesis of Bacillus cereus infections, Microbes Infect., 2000, 2(2), 189–198, DOI:10.1016/S1286-4579(00)00269-0.
- (a) P. A. Wayne, National committee for Clinical Laboratory Standards, 1997, Approved Standard M7–A4; (b) P. A. Wayne, National committee for Clinical Laboratory Standards, 1997, Approved Standard M27.
- J. R. Zgoda and J. R. Porter, A convenient micro dilution method for screening natural products against bacteria and fungi. Pharmaceutical, Biology, 2001, 39(3), 221–225 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All the 1H & 13C NMR and mass spectra of 4a–n are available. See DOI: 10.1039/c5ra11094g |
| This journal is © The Royal Society of Chemistry 2015 |