
Palladium(ii) complexes containing ONO tridentate hydrazone for Suzuki–Miyaura coupling of aryl chlorides in aqueous-organic media†
Abstract
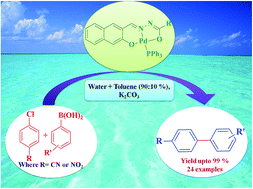
Facile synthesis of three new palladium(II) complexes bearing heterocyclic hydrazone ligands are presented along with their structural characterization using IR, 1H and 13C NMR spectra. Molecular structures of the complexes determined by single-crystal XRD revealed a distorted square-planar geometry around the metal ion to which the hydrazone was attached in a tridentate fashion. Catalytic activity of these complexes tested towards the Suzuki–Miyaura cross coupling reaction of substituted aryl boronic acids with aryl chlorides in a water–toluene system (90 : 10%) without using any promoting additives or phase transfer agents, proved that they are highly active with 0.01 mol% loading under optimized conditions to afford 99% yield of the coupled product. Effects of temperature, solvent and base on the cross-coupling reaction were carried out as well. These complexes showed significant catalytic activity up to five cycles.
 Please wait while we load your content...
Please wait while we load your content...

