
Polymer nanocomposites using click chemistry: novel materials for hydrogen peroxide vapor sensors†
Abstract
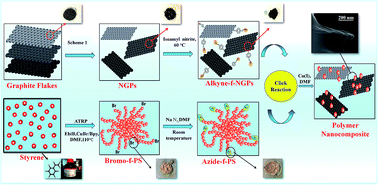
Herein, we report the synthesis of nanocomposites (NCs) prepared by azide-functionalized polystyrene (Az-f-PS) coupled with alkyne-functionalized nanographite platelets (Alk-f-NGPs) by using copper(I) catalyzed azide–alkyne click chemistry. Linear polystyrene (PS) was functionalized with an azide moiety after bromine-termination of PS through atom transfer radical polymerization (ATRP) using CuBr/2,2′-bipyridyl as a catalyst. The nanographite platelets (NGPs) were formed from graphite flakes through intercalation followed by functionalization with an alkyne moiety. The structure and micromorphology of the prepared NCs were confirmed by NMR, FTIR, Raman, XRD, TGA, SEM, TEM and AFM techniques. The electrical properties of the click nanocomposites (C-NCs) were investigated and the C-NCs were found to posses the resistance of 1.08 × 108 Ω at 1 wt% NGPs loading. The C-NCs with fast response (∼3 s), rapid recovery (∼60 s) and excellent repeatability at room temperature provide novel materials for chemiresistive sensors for detection of H2O2 vapors.
 Please wait while we load your content...
Please wait while we load your content...

