
Recent advances in biological applications of cage metal complexes
Yan Z. Voloshin*,
Valentin V. Novikov and
Yulia V. Nelyubina
Nesmeyanov Institute of the Organoelement Compounds, Russian Academy of Sciences, Moscow, Russia. E-mail: voloshin@ineos.ac.ru
First published on 11th August 2015
Abstract
This review highlights advances in biochemical and medical applications of cage metal complexes (clathrochelates) and related polyhedral compounds as rigid scaffolds for macromolecular binding (topological drugs), in radiation therapy and diagnostics, and as paramagnetic probes for NMR and MRI applications.
1. Introduction
Modern technologies heavily rely on knowledge from different fields of science. Today, novel pharmaceuticals, diagnostic methods and materials, such as biological/non-biological constructs,1 are constantly being produced at the crossroads of biology, materials engineering, biochemistry and synthetic chemistry. Until recently, the main efforts of medicinal chemists have been focused on creating new potent drugs within the realm of organic chemistry; however, over the last decade, the need for pharmaceuticals with improved potency and selectivity has shifted the attention of some researchers towards metal complexes.2 The advantages they provide over purely organic compounds are structural diversity,3 simple syntheses and specific characteristics, such as redox,4 optical,5 catalytic,6 radioactive7 and magnetic,8 induced by a metal ion.Whereas some medicinal applications of metal complexes, e.g. catalytic transformations in living organisms,9 require the metal ion to be easily accessible by the biological environment, other approaches may benefit from its total isolation, which may help to ensure stability of a biologically-active complex by avoiding its decomposition, transmetallation or unwanted coordination. An intuitive strategy in this case is to bury the metal ion inside a three-dimensional ligand, as in cage complexes (clathrochelates10). Encapsulation of a transition metal ion leads to complexes with high chemical stability and unusual properties that can be used as molecular scaffolds for new (photo)electronic devices (molecular switches,11 magnetic12 and electrochromic13 materials), molecular machines,14 electrocatalysts for hydrogen production,15 metallomacrocycles,16 metal-organic frameworks16b and coordination capsules.17
Biological applications of clathrochelates have been covered previously in 1996 (ref. 18) and 2007.19 This short review summarizes the progress that has been made in this area since then. First, biological aspects of clathrochelates will be addressed for which the role of the metal ion is purely structural, so all their activity is due to the caging ligand. Then applications will be highlighted that take advantage of the encapsulated metal ion itself: those include the use of clathrochelates as radioactive and magnetic probes for diagnostics and therapy.
2. Topological drugs: cage metal complexes as rigid scaffolds for macromolecular binding
The most biologically active agents bind to biological targets by either covalent bonds or supramolecular interactions; their selectivity and specificity determine the performance of these agents in drug therapy. Modern trends in drug development have led to the discovery of new biological targets, hidden allosteric sites (that is, compounds residing in special allosteric centres far from the active site of an enzyme causing a change in its properties) and interfaces of macromolecular interactions.The recent concept of “allosteric drugs”20 implies the presence of hidden allosteric sites in nearly all the existing proteins, thus offering opportunities for developing agents that inhibit them as shown in Scheme 1b. These sites are usually not known, and their choice depends heavily on the ability to create artificial molecules that are topologically complementary to them. Another modern approach in drug therapy is targeting protein–protein interactions; given a rather extensive surface of macromolecular interfaces, the modulation of these interactions requires relatively small but bulky inhibitor molecules of a suitable size (Scheme 1a).
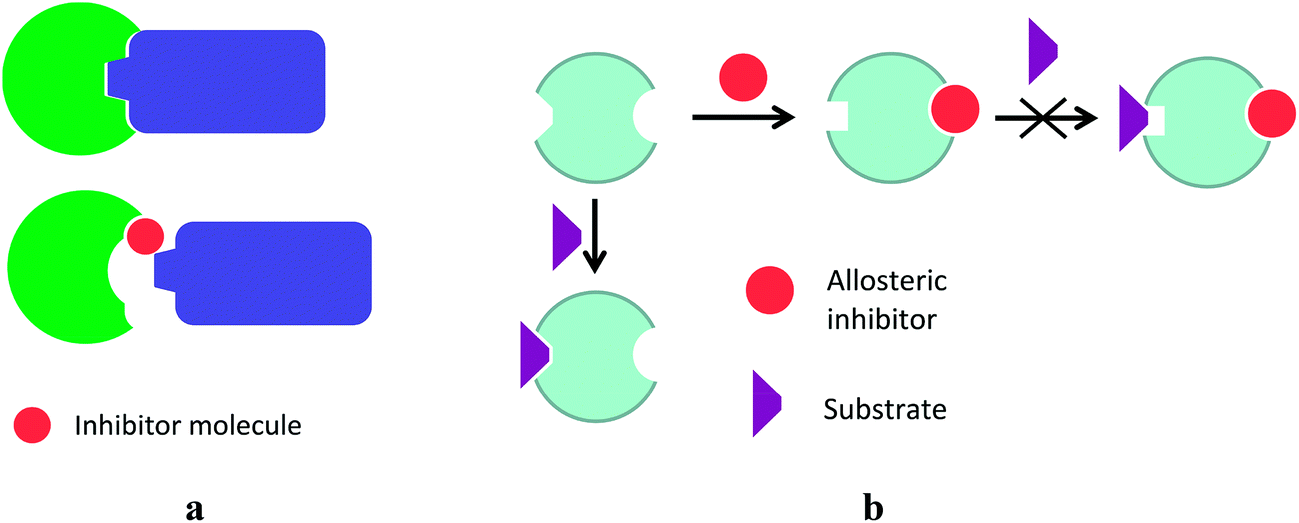 | ||
| Scheme 1 Inhibition of macromolecular protein–protein interactions by interface (a) and allosteric (b) inhibitors. | ||
These new biological targets demand new guest molecules with a tunable geometry and extensive surface.21 One concept,22 for which the term “topological drugs” has been coined,21 suggests the use of rigid three-dimensional species that form multicentered supramolecular interactions in the vacant cavities of protein macromolecules and their complexes. Among polyhedral and cage compounds (Fig. 1), transition metal clathrochelates,10 fullerenes,23 carboranes and metallacarboranes24 are large and bulky enough to ensure strong multicentered supramolecular binding by van der Waals interactions with large protein molecules. The design of topological drugs paves the way towards new antitumor and antiviral drug candidates; it is also possible that drug resistance is less likely to develop in these cases, if it is caused by the enzymatic deactivation of the active compound.
 | ||
| Fig. 1 Polyhedral and cage metal compounds. | ||
Clathrochelate-based xenobiotics, which have neither natural analogs nor structural similarities to biological molecules, are of particular interest. Examples of xenobiotics among polyhedral compounds are derivatives of the adamantane carbocycle (Fig. 1) widely used in drug therapy of Parkinson’s and other human viral, dermatological and psychical diseases. The activity of these compounds is usually attributed to the bulky carbocyclic polyhedron that allows them to be included into the lipid bilayers of biological membranes and to interact with biological targets such as the hydrophobic parts of proteins. Therefore, the therapeutic effects of these diamonoids are governed by their geometric complementarity to the corresponding receptors.25
Clathrochelates10 are topological analogs of the above polyhedral compounds. The applicability of these chemically robust cage complexes, in particular, polyazomethine macrobicyclic tris-dioximates, as possible drug candidates is also due to their synthetic availability and structural diversity; apical (the green balls in Fig. 1) and ribbed (the blue balls in Fig. 1) functionalization of clathrochelates can be easily performed using well-known organic reactions with commercially available chemical reagents. A typical synthetic pathway to metal clathrochelates (Scheme 2) is via template condensation of α-dioxime with a Lewis-acidic trigonal boron compound and a metal salt.10
 | ||
| Scheme 2 An exemplary synthetic pathway to transition metal clathrochelates. | ||
Various functionalizing substituents may be introduced into the ribbed fragments of a clathrochelate by N,O,S,P,C–nucleophilic substitution of halogen atoms in halogenoclathrochelates (Scheme 3); this can produce mono- and triribbed-functionalized cage complexes from mono-,26 di-27 and hexa-28 substituted halogenoclathrochelates.
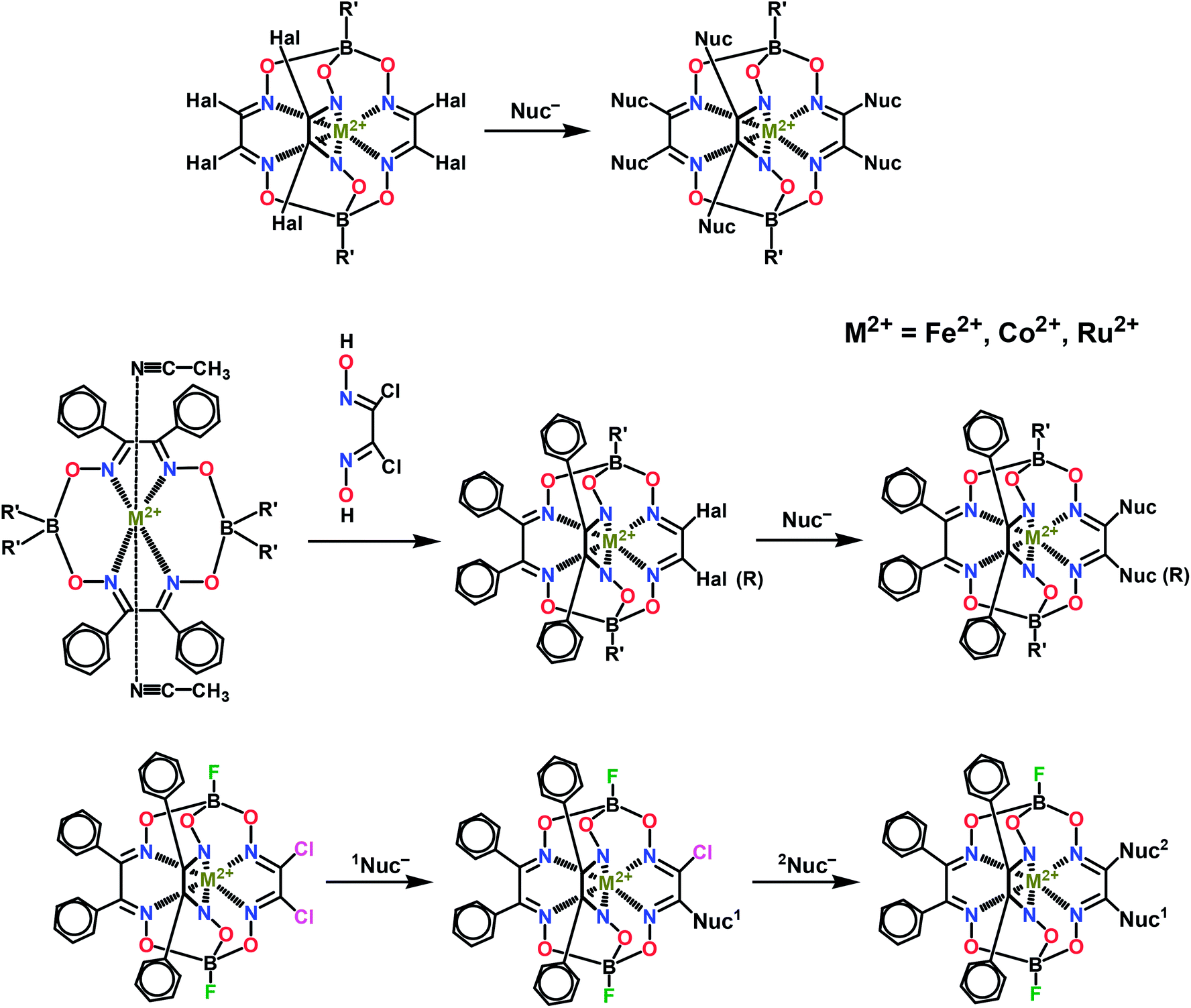 | ||
| Scheme 3 Functionalization of transition metal clathrochelates. | ||
The possibility to “build” a cage complex from a single center in all directions simultaneously (Fig. 1), combined with a wide range of available functionalizing groups (including pharmacophore fragments), allows designing active compounds with shapes closely matching macromolecular surfaces of a very complex topology (such as protein–protein and protein–DNA interaction interfaces) to ensure the effective binding of biological macromolecules that affect the structure and functions thereof.21
Binding proteins
Among a series of iron(II) mono- and bis-clathrochelates (Scheme 4), the compounds 7, 8 and 18 have been tested for binding to bovine serum albumin (BSA), β-lactoglobulin, lysozyme, and insulin29 using steady-state and time-resolved fluorescence spectroscopies. Quenching of the protein’s fluorescence and the decrease in its excited state lifetime has been observed only in the case of BSA, which was evidence for the formation of strong assemblies by the clathrochelates with serum albumins and not with other globular proteins.29 | ||
| Scheme 4 Iron(II) mono- and bis-clathrochelates with reported biological activity. | ||
The binding of a metal clathrochelate, zinc(II) diaminosarcophaginate 26 (Scheme 5), to bovine and human serum albumins (BSA and HSA) has been explored30 under simulated physiological conditions. The observations from fluorescence spectroscopy have shown this cage complex to effectively quench the intrinsic fluorescence of BSA and HSA via static quenching processes. Binding sites and binding constants, thus identified and supported by molecular docking, have suggested that the binding process occurred via hydrogen bonding and van der Waals interactions between the clathrochelate and the BSA or HAS species.
 | ||
| Scheme 5 Transition metal sarcophaginates with reported biological activity. | ||
The same experimental and theoretical approaches have been used31 for studying the binding of manganese(II) sarcophaginate 27 (Scheme 5) to these proteins in vitro. FT-IR data obtained have shown that the conformations of the proteins and their microenvironments changed in the presence of 27; the latter strongly quenched their intrinsic fluorescence by static quenching. Hydrogen bonding and weak van der Waals interactions have been found to play a key role in stabilizing the resulting clathrochelate–protein supramolecular assemblies. Note that the initial diaminosarcophagine (a caging ligand with no metal ion trapped inside) has also exhibited good binding properties towards HSA and BSA with relatively high binding constants; according to FT-IR data, the secondary protein structures32 have slightly changed upon this binding.
A library of iron(II) tris-dioximate clathrochelates has been screened for inhibitory activity against HIV protease in silico.33 The binding of the lead compound has been observed to occur in an unusual mode, with the cage complex located at the periphery of the protein’s cavity rather than at the catalytic aspartate diad, as was expected.
Targeting protein–DNA interactions
The first example of efficient transcription inhibition by a transition metal clathrochelate has been reported for a model in vitro system based on a T7 RNA polymerase (T7 RNAP), a small single-subunit polymerase that was often used as a convenient and reliable model in the search for transcription inhibitors.34 The in vitro testing of iron(II) clathrochelates 1–8 (Scheme 4) in the transcription assay has demonstrated structure- and concentration-dependent inhibition by most of them. Among these complexes, two monoribbed-functionalized iron(II) clathrochelates 7 and 8, the derivatives of meta- and para-mercaptobenzoic acids, have been found to suppress the transcription of T7 RNA in a submicromolar range. Heterofunctionalized iron(II) clathrochelates 14–16 with good ADMET properties,26c e.g. sufficient water solubility, membrane permeability and low toxicity, have also demonstrated a good inhibitory activity in the low- and submicromolar concentration range.A series of cobalt(II) bis(1,2-dicarbollides) have been recognized as effective HIV protease inhibitors.35 To increase their activity even further, it was proposed to join two identical polyhedral fragments by a flexible linker;36 the resulting construct displayed an activity that was twice as high. A similar approach employing close and relatively rigid joining of two identical clathrochelate frameworks (Scheme 6) has been used to obtain a clathrochelate-based inhibitor having a vastly expanded binding surface with a relatively low increase in conformational flexibility.37 The C–C conjugated bis-clathrochelates thus obtained have inhibited the transcription in the T7 RNAP system extremely efficiently, with values of IC50 below the submicromolar range; this places them among the most potent metal-based transcription inhibitors to date.37a Although they have limited torsion mobility around the C–C bond that links the two cage frameworks, these bis-clathrochelates are able to alter their geometry to match the shape of the binding site, while keeping the number of conformational states low to ensure higher selectivity.37a
 | ||
| Scheme 6 Synthesis of C–C conjugated iron(II) bis-clathrochelates. | ||
Binding of the above iron(II) mono- and bis-clathrochelates (Scheme 4) to T7 RNAP has been found in silico26c,34a,37a to occur via the occupation of the same region of a macromolecular complex – the transcriptional bubble – and involved intermolecular contacts with protein residues as well as with DNA and RNA. The clathrochelate molecule resided in the pocket formed by the surfaces of DNA and RNA and completed by the two structural elements of T7 RNAP that interacted with its phenyl substituents, thus trapping the clathrochelate molecule inside. The inhibition of the transcription in this system was ensured by blocking further strand separation via the binding of the clathrochelate in the transcription bubble to give a quaternary complex that was able interfere with the translocation step of this reaction. This has been confirmed by preincubation of the lead compound with DNA, with T7 RNAP and with their mixture;34a the experimental data have been in good agreement with the theoretical results from molecular docking, all suggesting the DNA–RNAP complex (or even a DNA–RNA–RNAP ternary complex) as a target for these clathrochelate-based inhibitors. The observed mode of inhibition by the mono- and bis-clathrochelates that involved a binding pocket formed by three interconnected macromolecules of the transcription complex has been attributed34a,37a to their rather unique structural features: these complexes are large enough to ensure efficient binding mostly by van der Waals interactions; this binding mode seems to be less probable for small and planar molecules.
Targeting protein–protein interactions
Transition metal clathrochelates have only recently emerged as anti-fibrillogenic agents38 to fight neurodegenerative disorders (such as Alzheimer’s, Parkinson’s and Creutzfeldt–Jakob’s), type II diabetes and amyloidosis, all caused by the deposition of insoluble protein β-pleated formations (aggregates or amyloid fibrils) in the cells and extracellular space of various organs or tissues. Addition of the iron(II) mono- and bis-clathrochelates 7, 8, and 17 (Scheme 4) to an in vitro system with insulin has significantly changed the kinetics of insulin fibrillization, reduced the amount of fibrils formed (up to 70%), and caused a decrease in their diameter to 3–8 nm from the 5–12 nm typical for free insulin fibrils. These complexes also prevented the lateral aggregation of mature fibrils and the formation of superfibrillar clusters. The highest inhibitory activity has been observed for the monoclathrochelate derivative of the meta-mercaptobenzoic acid 7: its efficient inhibitory concentration IC50 was as low as 16 ± 2 μM.38Targeting DNA–DNA interactions
A transition metal clathrochelate, cobalt(III) sepulchrate 28 (Scheme 5), has been reported to promote DNA condensation39 by forming macromolecular complexes with several molecules of DNA. This clathrochelate effectively precipitated DNA at high concentrations, thus emerging as a much more efficient condensation agent than its non-macrocyclic analogues. Although electrostatic interactions are the main driving force for the binding of multivalent complex cations (such as a sepulchrate cation) to DNA in a solution, the process of DNA condensation also depends on their structure; supramolecular recognition and consequent DNA condensation by such cations are affected by changes in their size, chemical composition and surface.393. Cage complexes for radiation therapy and diagnostics
Nuclear medicine uses radiation to treat specific organs or to provide diagnostic information on how they function. A recent development of nuclear medicine, Positron Emission Tomography (PET),40 is a more precise functional imaging technique used mostly for identifying medical conditions (including cancers, heart disease and brain disorders) or following the progress of their treatment.Among transition metals that can be buried in a boron-based macrobicyclic ligand to produce a clathrochelate, those that provide radioisotopes useful for this purpose include copper and cobalt. Isotopes of copper 60Cu, 61Cu, 62Cu, and 64Cu are positron emitters with half-lives ranging from relatively short (9.74 minutes for 62Cu) to relatively long (12.7 hours for 64Cu).40b,41 The long half-life of 64Cu allows it to be used in PET imaging as well as for radiotherapy; the selectivity and target delivery of the 64Cu isotope to a biological organ are very important for its radiopharmaceutical applications.
Biodistribution of 64Cu2+ complexes is governed by their stability, size, shape, charge, lipophilicity, and redox properties, and further selectivity can be achieved using vector substituents that bind to specific molecular targets in vivo.40b Among copper(II) clathrochelates, sarcophaginates functionalized by biologically relevant groups have been recognized as promising candidates for future PET imaging agents.42 Scheme 7 shows a synthetic pathway to a copper(II) sarcophaginate 29 which can be turned into a sarcophagine 30 specifically designed43 for binding to peptides. Functionalization of polyamine macrobicyclic ligands (such as sarcophagines) is a real challenge, as they have eight reactive centers (two apical primary and six ribbed secondary amino groups) resulting in a poorly separable mixture of clathrochelate products.
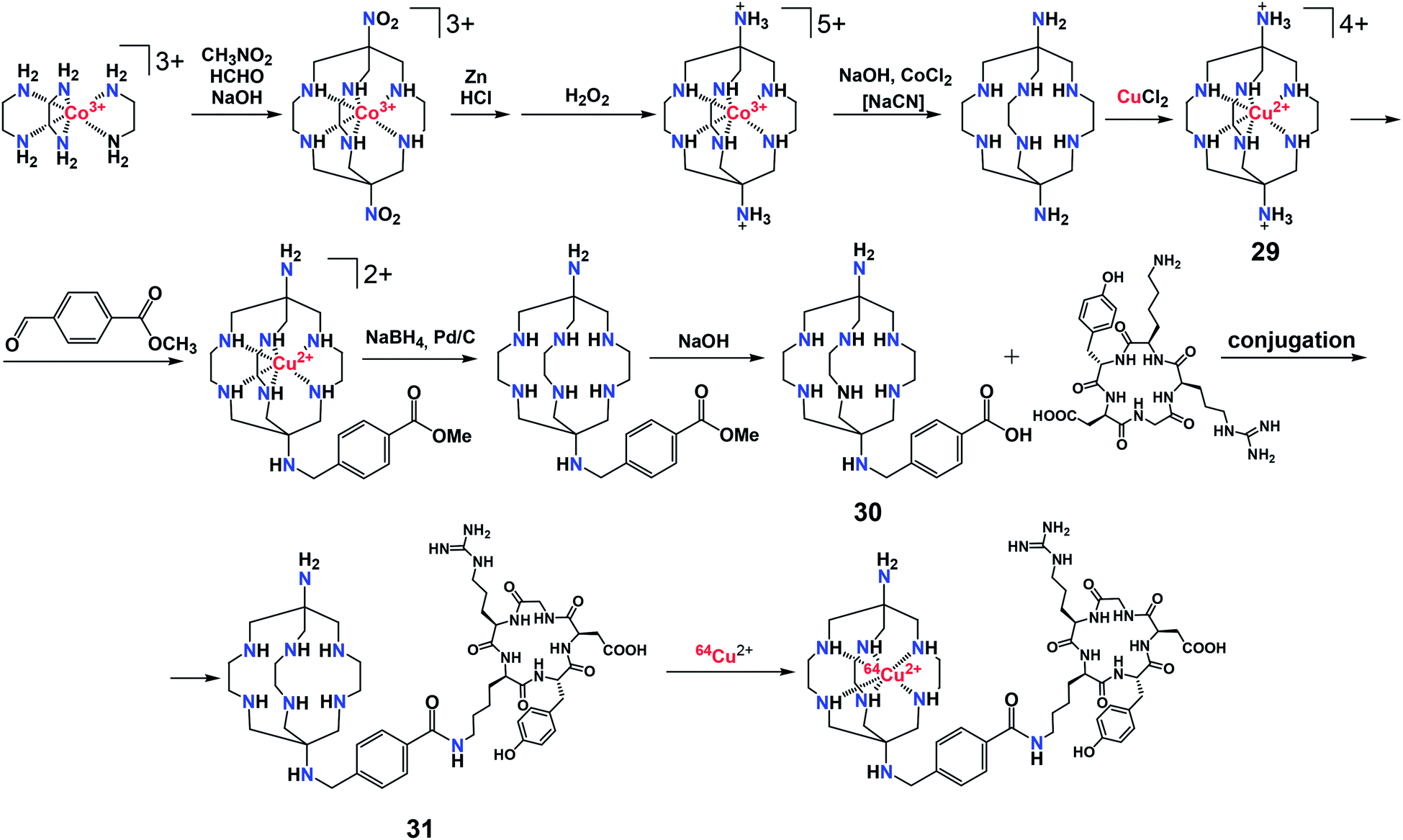 | ||
| Scheme 7 Synthesis of a copper(II) complex with sarcophagine 31. | ||
In the copper(II) sarcophaginate 29, the donor secondary amino groups are deactivated by their coordination to the encapsulated metal ion. The demetallation of this intermediate complex gave a monofunctionalized aminosarcophagine 30, which then underwent condensation with a cyclic peptide. The resulting sarcophagine 31 allowed a targeted delivery of its 64Cu2+ complex.43 The latter may also be synthesized with no need for the isolation of an intermediate copper(II) sarcophaginate 29.44 In this case, the intermediate sarcophagine to be further functionalized can be obtained from a corresponding cobalt(III) cage complex either by its demetallation followed by base hydrolysis of the free ligand or by its base hydrolysis followed by demetallation (Scheme 8).
 | ||
| Scheme 8 Synthesis of a carboxyl-terminated sarcophagine 30. | ||
An analogous bifunctionalized sarcophagine 32 (Scheme 9) has been synthesized45 by the direct alkylation of its diaminosarcophagine precursor with 4-bromobenzoic acid. Its condensation with the corresponding peptide gave a sarcophagine 33, the copper 64Cu2+ complex of which showed an uptake by tumor cells exceeding that for the sarcophaginate of 30. Note that a 68Ga3+-encapsulating analogue46 turned out to be a promising non-invasive PET imaging agent.
 | ||
| Scheme 9 Synthesis of a sarcophagine 32 and its peptide conjugate 33. | ||
Mono- and bifunctionalized copper(II) sarcophaginates 34–36 (Scheme 10) with di- or trimethylene linkers and with terminal reactive carboxylic groups47 have been obtained from octaamine precursors; their further functionalization with various peptides has allowed 64Cu2+ sarcophaginates to be obtained that were stable for a long time under the conditions of in vivo experiments.48 Another sarcophaginate-based bioconjugate 38 (Scheme 11) with the encapsulated 64Cu2+ ion, which performed well in both in vitro and in vivo experiments,49 has been synthesized via 1,3-dipolar cycloaddition of a dibenzocyclooctyne-functionalized 64Cu2+ sarcophaginate 37 with an azide-containing cyclic peptide.
 | ||
| Scheme 10 Synthesis of mono- and bifunctionalized copper(II) sarcophaginates 34–36. | ||
 | ||
| Scheme 11 Synthesis of a sarcophaginate-based bioconjugate 38. | ||
Copper(II) sarcophaginates 41 and 42, specifically designed for conjugation with antibodies,50 have been obtained from isothiocyanate-containing sarcophagines 39 and 40 (Scheme 12). According to in vivo biodistribution and PET imaging data, their 64Cu-labelled radioimmunoconjugates with a HER2/neu-targeting antibody have shown high stability under physiological conditions with a high and selective uptake in a HER2-positive cancer cell line. Cage complexes of homo- and heteromultifunctionalized sarcophagines 43–46 (Scheme 13) with the encapsulated 64Cu2+ ion have also demonstrated good in vitro and in vivo stability,51 making these multifunctional chelators versatile molecular platforms for multivalent/multimodality probes for both imaging and therapy.
 | ||
| Scheme 12 Pathway to and functionalization of isothiocyanate-containing sarcophaginates 41 and 42. | ||
 | ||
| Scheme 13 Homo- and heterofunctionalized sarcophagine chelators for the 64Cu2+ cation. | ||
A 64Cu-labelled complex of a dipeptide sarcophagine 47 (Scheme 13), synthesized from a diazide-containing sarcophagine and a corresponding alkyne-containing peptide, has been used52 for the microPET imaging of the receptor CD13 (a tumor vasculature biomarker) expressed in vitro and in vivo in living mice; it displayed a good binding affinity and CD13 specificity with given tumor cells and an excellent tumor uptake. Similarly, 64Cu2+ cage complexes of exendin-4-functionalized sarcophagines 48 and 49, having high kidney radioactivity levels,53 showed a persistent and specific uptake in a given insulinoma model; an increased tumor uptake in vivo has been also observed for 49. These sarcophagines and their 64Cu2+ complexes are, therefore, promising agents for 1-targeted PET imaging of the glucagon-like peptide. The receptor of this peptide with an elevated expression profile in pancreatic islets, insulinoma, and cardiovascular system53 and its accurate visualization and quantification of β-cell mass are critical for the improved understanding, diagnosis, and treatment of type 1 diabetes and insulinoma.
A 64Cu-labelled sarcophaginate 50 (Scheme 13), which contained a near-infrared fluorescent dye fragment and a peptide’s exendin-4 moiety, has appeared to be suitable for the bimodal PET–fluorescence imaging of the glucagon-like peptide 1 expression in the pancreas and in pancreatic islet cell tumors.54 It has demonstrated good performance in vivo and ex vivo, showing small xenografts with PET and pancreatic β-cell mass by phosphor autoradiography. Its fluorescence has also allowed the detection of individual pancreatic islets, thus confirming specific binding to a target peptide with sensitivity surpassing that of a radioactive label. These dual imaging probes54 can be used for the diagnosis of primary growths and metastases and for the detection of tumor margins, infiltrative growth and residual tumor cells. A heterofunctionalized sarcophaginate 51 (ref. 55) has also been proposed as a candidate for the further design of various tumor-targeted dual-modality imaging probes.
Novel 64Cu-labeled radiopharmaceuticals have been obtained from bifunctionalized sarcophagines 52 and 53 and their peptide-containing derivatives 54 and 55 (Scheme 14), which exhibited high tumor uptake and tumor-to-normal tissue ratios.56 Triamidetriamine macrobicyclic and macrotricyclic ligands 56 and 57 for the delivery of the radioactive 64Cu2+ ion (Scheme 15) have been synthesized57 using a non-template synthetic strategy; the resulting 64Cu2+ cage complex of 56 showed no signs of decomposition when incubated with L-cysteine and was rather stable in the presence of L-histidine as a concurrent N-donor ligand. The corresponding bifunctional sarcophagine has been conjugated with a tumor-targeting peptide Tyr3-octreotate to give a sarcophagine 58. The corresponding 64Cu2+ complex had high selectivity for tumor cells expressing somatostatin receptor 2 (sstr2) and an excellent uptake in sstr2-positive tumors, as shown both by in vitro and in vivo studies.58 If labelled with 64Cu2+ and 67Cu2+ ions, the cage complex of 58 can be suitable for the combined imaging and therapeutic treatment of neuroendocrine tumors.
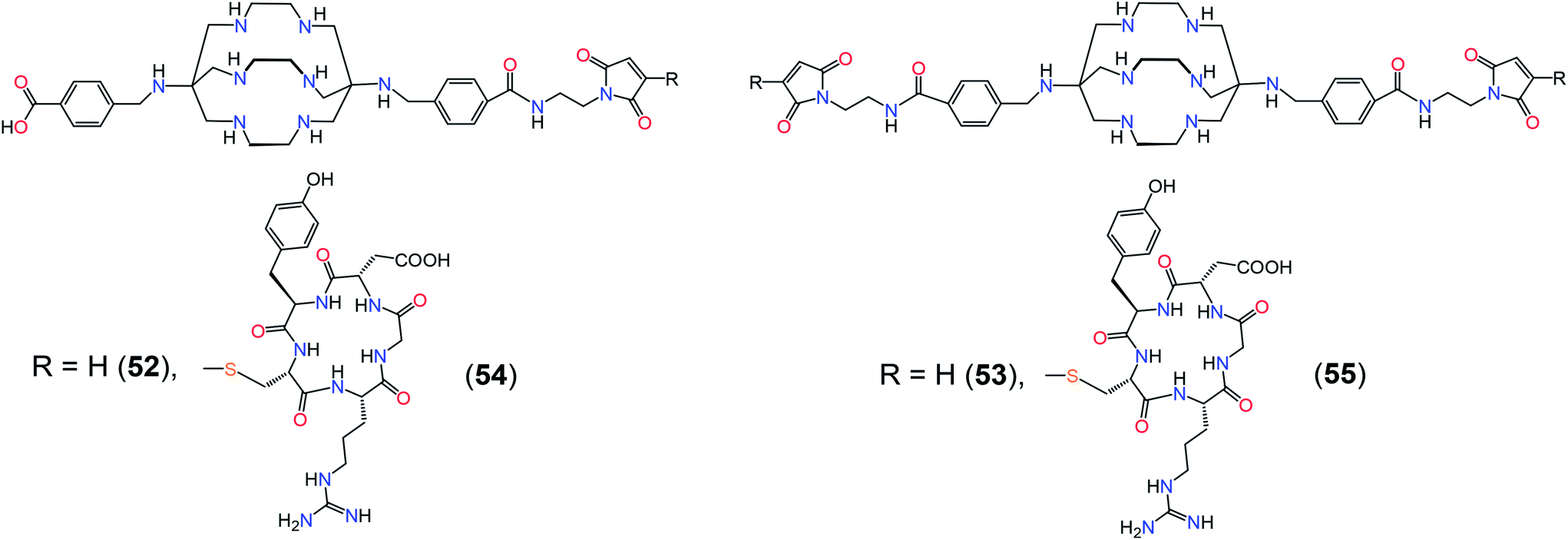 | ||
| Scheme 14 Bifunctionalized sarcophagines 52 and 53 and their peptide-containing derivatives 54 and 55. | ||
 | ||
| Scheme 15 Macrobi- and macrotricyclic ligands 56 and 57 and a peptide-functionalized sarcophagine 58. | ||
Conjugates of copper(II) sarcophaginates with antibodies, formed by suitable linkers (Scheme 16), have been proposed for the targeted delivery of the 64Cu2+ ions to a given biological system.59 For example, the conjugate of a single-chain antibody with a monofunctionalized 64Cu2+ sarcophaginate (Scheme 17) has been used for the diagnostic PET imaging of activated platelets.60 It accumulated in an injured vessel with a high and specific uptake, thus paving the way towards highly sensitive in vivo detection of activated platelets and early diagnosis of acute thrombotic events.60
 | ||
| Scheme 16 Conjugation of 64Cu2+ sarcophaginates with antibodies. | ||
 | ||
| Scheme 17 Conjugate of a single-chain antibody with a monofunctionalized sarcophaginate. | ||
Similarly, the diagnostic potential for carotid artery thrombosis using PET has been demonstrated by a conjugate (Scheme 18) obtained via enzyme-mediated site-specific bioconjugation of a 64Cu2+ sarcophaginate with a single-chain antibody.61 A fragment of this antibody targeted the ligand-induced binding sites on a glycoprotein receptor GPIIb/IIIa found on the activate platelets. The conjugate radiolabeled with the positron-emitting 64Cu ions has been shown to selectively bind these platelets in an in vivo model.
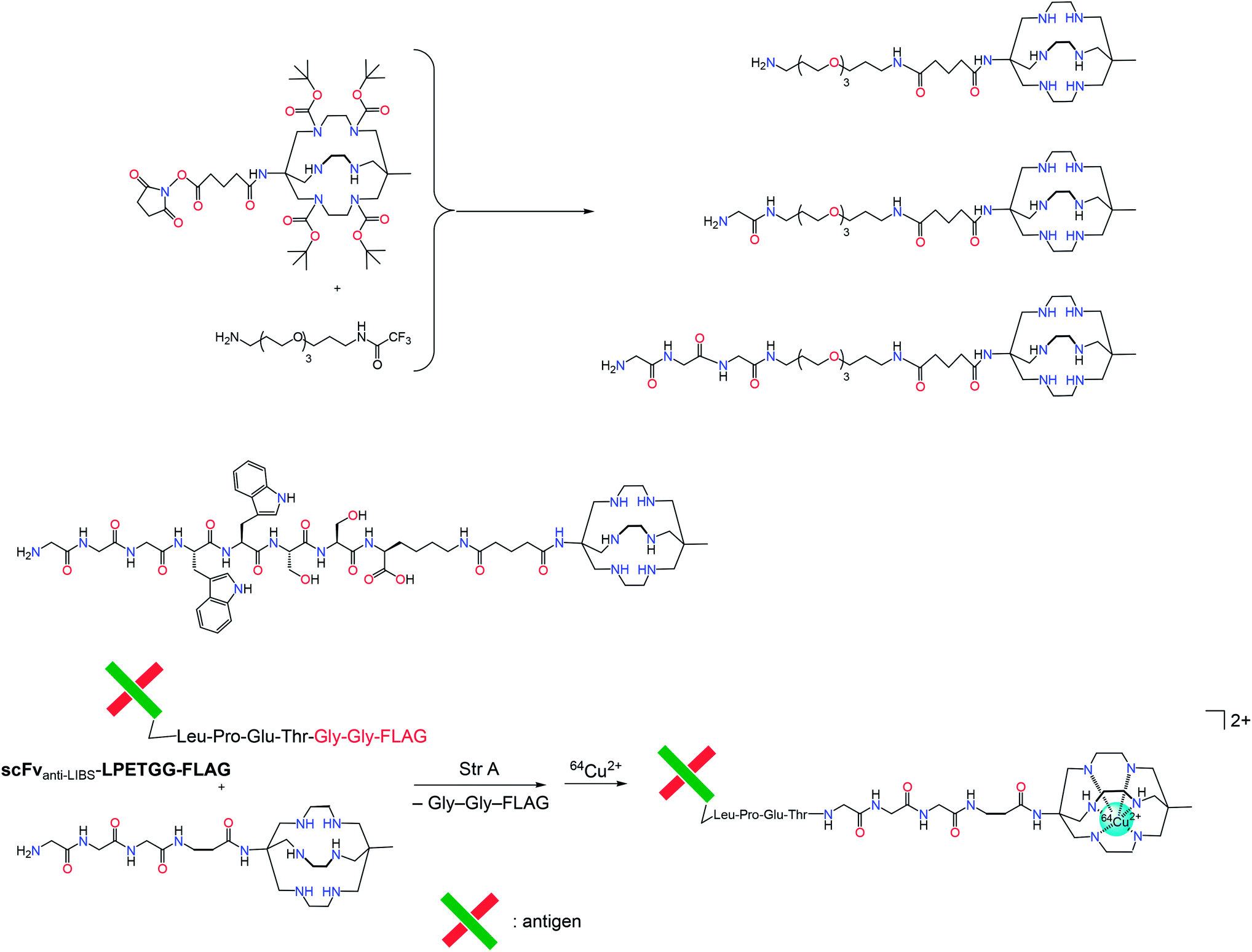 | ||
| Scheme 18 Monofunctionalized sarcophagines and their conjugates with a single-chain antibody for 64Cu2+ delivery. | ||
A series of conjugates formed by eight different bifunctional sarcophagines (Scheme 19) has been probed for radiolabeling efficiency, in vitro stability, and biodistribution.62 Among those, a 64Cu2+-label sarcophaginate 59 conjugated with an anti-CD20 antibody has appeared to be extremely stable in vivo and to achieve high specific activity under very dilute conditions. A peptide-functionalized 64Cu2+ sarcophaginate 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 63 has also turned out to be an efficient probe for non-invasive PET imaging, in this case, of tumor-associated α2β1 expression, thus being a useful tool for identifying prostate cancer.
63 has also turned out to be an efficient probe for non-invasive PET imaging, in this case, of tumor-associated α2β1 expression, thus being a useful tool for identifying prostate cancer.
 | ||
| Scheme 19 64Cu2+-labelled conjugate 59 and a peptide-functionalized sarcophaginate 60. | ||
Heterobifunctionalized sarcophagines have also found use in the design of long-circulating nanotherapeutics.64 Nanoparticles formed by polypeptides modified by these 64Cu2+ cage complexes via site-specific conjugation retained their activity in vivo over several days, and their kinetics could be followed by microPET imaging.64
4. Cage complexes as paramagnetic probes for NMR and MRI applications
Nuclear magnetic resonance (NMR) and magnetic resonance imaging (MRI) have a well-deserved reputation as the most powerful techniques for studying biological systems. NMR spectroscopy is one of the main sources of information about their structural features; despite being limited by the size of the biomacromolecule under study, it nevertheless allows the determination of its native conformation in a solution, thus ensuring continuing progress in this field.One of the possible approaches to increase the accuracy of the structural information thus obtained is to use paramagnetic probes. Although most chemical applications of NMR spectroscopy consider the presence of paramagnetic species an unfortunate complication, both MRI and biological NMR often benefit from the effects they exert on the magnetic properties of a system.65 Paramagnetic labels, i.e. coordination compounds with paramagnetic transition metal or lanthanide ions covalently attached to the biological macromolecules, are gaining popularity as probes for NMR spectroscopy to help unravel insightful details of structure and dynamics of proteins and their complexes. Some well-established applications of paramagnetic tags include NMR structure determination in solution,66 solid-state NMR crystallography,67 and characterization of otherwise inaccessible low-populated conformational states.68
Paramagnetic probes make use of the interactions of the spin of an unpaired electron with the magnetic moments of the surrounding nuclei. An important feature of a paramagnetic ion is the electron relaxation time that is significantly shorter than the nuclear relaxation time. Slow electronic relaxation induces fast relaxation of nearby nuclei, thus resulting in a significant broadening of their NMR signals (so-called paramagnetic relaxation enhancement, PRE). Another feature is the magnetic anisotropy of a paramagnetic ion, which may result in very large paramagnetic shifts (so-called pseudocontact shifts, PCSs) of the NMR signals of the surrounding nuclei as well as in the partial orientation of the paramagnetic probes in the magnetic field giving rise to residual dipolar couplings (RDCs). As all these effects depend on the coordinates of the corresponding nuclei in the molecular frame of a paramagnetic ion found in a relaxation or a shift agent, the data they provide contain detailed information about the structure and dynamics of a biomolecule.
MRI, which is unquestionably one of the most informative diagnostic techniques in modern medicine, also relies heavily on paramagnetic probes. Differentiation of tissues from different organs is achieved by using inherent differences between the relaxation times of water protons in different biological environments; this contrast may be enhanced even further by administering contrast (relaxation) agents, usually chelate complexes of gadolinium. A recently recognized adverse effect of gadolinium salts on certain groups of renally compromised patients, that is, causing nephrogenic systemic fibrosis,69 triggered an ongoing search for new contrast agents that were immune to transmetallation of the chelated gadolinium70 or did not contain it at all.
Both paramagnetic shift and contrast agents require the highest possible stability of the paramagnetic species against the loss of the metal ion, combined with their small overall size. Shift agents should also have a large magnetic anisotropy to be effective, and contrast agents require the paramagnetic ion to have access to bulk water. All these seemingly conflicting conditions can be met if the metal ion is encapsulated in a three-dimensional cage; the most prominent examples are gadofullerenes,71 in which the gadolinium cation or a charged metal-containing cluster is buried in a carbon cage.72 In contrast to chelate-based contrast agents, it is almost impossible to lose the metal ion in this case, as it is trapped inside an impervious fullerene cage. Although usually such encapsulation would decrease the relaxivity owing to the absence of the direct water–metal coordination that is necessary for inner-sphere relaxation, gadofullerenes have shown surprisingly high relaxivities73 resulting from the formation of nanocluster aggregates74 with large rotation correlation times. This makes the gadofullerenes more similar to nanoparticle-based contrast agents75 rather than to molecular probes with all the associated limitations of the former, including limited ability to cross the blood–brain barrier for brain tumor imaging. As the encapsulation in the gadofullerenes does not occur via chelation, they are beyond the scope of this short review; an interested reader is referred to the excellent reviews on this topic.76
Clathrochelates also have a fully encapsulated metal ion, thus ensuring their stability; however, the only report on their use as relaxation agents dates back to 1992.77 For manganese(II), copper(II), and chromium(III) sarcophaginates (27, 75, and 76 from Scheme 5), a modest relaxivity in simple aqueous solutions was found, which dramatically increased upon the binding of the complex to a protein. This increase in relaxivity has been attributed to the larger rotation correlation time of the protein, so it had the same nature as in the gadofullerenes. Also being redox-active and synthetically available, the transition metal clathrochelates can be therefore considered as worthy rivals to the contrast agents based on gadofullerenes, with which they share chemical stability and high relaxivity.
In the field of paramagnetic labels, lanthanide ion complexes also account for most of the labels available to date,78 as they have an extremely large magnetic anisotropy, reasonably high chemical stability, and tunable (by the choice of a specific lanthanide ion) paramagnetic behavior. Magnetic properties of the central ion govern the changes in chemical shifts, the relaxation rates, and the weak alignment of a biomolecule in a magnetic field; all of those resulting in restraints useful for structure calculations.79 Their further advancement is, however, hampered by the high coordination number of lanthanide ions, the large size of the chelating probes, the formation of several diastereomers80 or conformational isomers81 with different magnetic properties, and by the net positive charge of the probe.82
Although 3d transition metal complexes have been the first to show residual dipolar couplings,83 they are rarely considered as shift agents, as they usually have a significantly lower magnetic anisotropy resulting in smaller paramagnetic pseudocontact shifts and a lower alignment ability. Additionally, these complexes are often not stable enough owing to the oxidation of the central metal ion or its undesirable coordination, and the delocalization of unpaired electrons over the ligand leads to an unwanted contact contribution to the paramagnetic shifts that makes the interpretation of the experimental results even more difficult. As a result, the use of transition metal complexes is mostly limited to assessing natural metal-binding sites84 or, in a chelated form, to their use as relaxation agents.85
Clathrochelates eliminate most of the limitations caused by chemical stability or unwanted coordination; however, a small (for low-spin cobalt(II) complexes86) or non-existent magnetic anisotropy precluded their use as shifting agents until recently. In 2010, it was found that high-spin cobalt(II) tris-dioximate clathrochelates87 with an unusual trigonal prismatic geometry had extremely large paramagnetic shifts in NMR spectra (Fig. 2) and a very large anisotropy of the magnetic susceptibility, reaching as high as 12.6 × 10−32 m3 at room temperature.12c Recently, the use of a tris-pyrazoloximate ligand allowed a cobalt(II) pseudo-clathrochelate88 to be obtained with an even larger magnetic anisotropy. The estimated value of 25 × 10−32 m3 not only surpassed that for most of the lanthanide-based paramagnetic tags but also appeared to be enough to result in single-ion magnet behavior.89 This shows the huge potential of the transition metal clathrochelates as new paramagnetic labels and contrast agents in biomolecular NMR and MRI, which, however, needs to be explored further before they can be placed into the toolkit of modern medicine.
 | ||
| Fig. 2 Paramagnetically-shifted 1H NMR spectra of high-spin cobalt(II) clathrochelates (a and b) and a hexadecylboron-capped pseudoclathrochelate (c). | ||
5. Conclusion
Metal complexes have long found use in many areas of material and medicinal sciences, yet new ones are still emerging every day. In this short review, we have briefly covered the recent progress in a fast developing area of the biological applications of a unique type of the complexes with an encapsulated metal ion, the clathrochelates. In contrast to many reported metal-containing therapeutic and diagnostic agents, for these cage compounds it is possible to clearly demarcate biologically relevant properties evoked by the encapsulated metal ion (such as radioactive and magnetic properties) and those governed by ligand-related size-and-shape features. While some of the reviewed complexes (e.g. radiotherapeutic agents) are on the verge of entering clinical trials, medicinal applications of others are still to emerge.Acknowledgements
The authors acknowledge the Russian Science Foundation (project 14-13-00724) for financial support.References
- (a) Z. J. Lesnikowski, Curr. Org. Chem., 2007, 11, 355–381 CrossRef CAS; (b) A. M. Chad and T. A. Taton, Nature, 2000, 405, 626–627 CrossRef PubMed; (c) A. S. Abd-El-Aziz, C. E. Carraher, C. U. Pittman, J. E. Sheats and M. Zeldin, Macromolecules Containing Metal and Metal-Like Elements, Biomedical Applications, Wiley-Interscience, 2004, vol. 3 Search PubMed.
- (a) S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC; (b) K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC; (c) N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC; (d) M. Dörr and E. Meggers, Curr. Opin. Chem. Biol., 2014, 19, 76–81 CrossRef PubMed.
- (a) E. Meggers, Chem. Commun., 2009, 1001–1010 RSC; (b) E. Meggers, Angew. Chem., Int. Ed., 2012, 51, 1–4 CrossRef PubMed.
- M. Corsini, F. Fabrizi de Biani and P. Zanello, Coord. Chem. Rev., 2006, 250, 1351–1372 CrossRef CAS PubMed.
- D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- (a) V. Amendola, Coord. Chem. Rev., 2006, 250, 1451–1470 CrossRef CAS PubMed; (b) P. S. Donnelly, Dalton Trans., 2011, 999–1010 RSC.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- Y. Z. Voloshin, N. A. Kostromina and R. Krämer, Clathrochelates: synthesis, structure and properties, Elsevier, Amsterdam, 2002 Search PubMed.
- S. Viswanathan, Y. Z. Voloshin, H. Radecka and J. Radecki, Electrochim. Acta, 2009, 54, 5431–5438 CrossRef CAS PubMed.
- (a) V. V. Novikov, I. V. Ananyev, A. A. Pavlov, M. V. Fedin, K. A. Lyssenko and Y. Z. Voloshin, J. Phys. Chem. Lett., 2014, 5, 496–500 CrossRef CAS PubMed; (b) M. Azarkh, L. Penkova, S. Kats, O. Varzatskii, Y. Voloshin and E. Groenen, J. Phys. Chem. Lett., 2014, 5, 886–889 CrossRef CAS PubMed; (c) V. V. Novikov, A. A. Pavlov, A. S. Belov, A. V. Vologzhanina, A. Savitsky and Y. Z. Voloshin, J. Phys. Chem. Lett., 2014, 5, 3799–3803 CrossRef CAS PubMed.
- A. B. Burdukov, M. A. Vershinin, N. V. Pervukhina, E. G. Boguslvaskii, I. V. Eltsov, L. A. Shundrin, S. L. Selector, A. V. Shokurov and Y. Z. Voloshin, Inorg. Chem. Commun., 2014, 44, 183–187 CrossRef CAS PubMed.
- (a) Y. Z. Voloshin, O. A. Varzatskii, M. Y. Antipin, S. V. Korobko, V. Y. Chernii, S. V. Volkov, V. I. Pehn’o and Z. A. Starikova, Inorg. Chem., 2005, 44, 822–824 CrossRef CAS; (b) J. R. Sabin, O. A. Varzatskii, Y. Z. Voloshin, Z. A. Starikova, V. V. Novikov and V. N. Nemykin, Inorg. Chem., 2012, 51, 8362–8372 CrossRef CAS PubMed.
- (a) O. Pantani, S. Naskar, R. Guillot, M. Millet, E. Anxolabéhère-Mallart and A. Aukauloo, Angew. Chem., Int. Ed., 2008, 47, 9948–9950 CrossRef CAS PubMed; (b) Y. Z. Voloshin, A. V. Dolganov, O. A. Varzatskii and Y. N. Bubnov, Chem. Commun., 2011, 47, 7737–7739 RSC; (c) M. T. D. Nguyen, M.-F. Charlot and A. Aukauloo, J. Phys. Chem. A, 2011, 115, 911–922 CrossRef CAS PubMed; (d) P. Millet, N. Mbemba, S. A. Grigoriev, V. N. Fateev, A. Aukauloo and C. Etiévant, Int. J. Hydrogen Energy, 2011, 36, 4134–4142 CrossRef CAS PubMed; (e) Y. Z. Voloshin, A. S. Belov, A. V. Vologzhanina, G. G. Aleksandrov, A. V. Dolganov, V. V. Novikov, O. A. Varzatskii and Y. N. Bubnov, Dalton Trans., 2012, 6078–6093 RSC; (f) E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 6104–6107 CrossRef PubMed; (g) A. V. Dolganov, A. S. Belov, V. V. Novikov, A. V. Vologzhanina, A. Mokhir, Y. N. Bubnov and Y. Z. Voloshin, Dalton Trans., 2013, 4373–4376 RSC; (h) I. G. Belaya, S. V. Svidlov, A. V. Dolganov, G. E. Zelinskii, T. V. Potapova, A. V. Vologzhanina, O. A. Varzatskii, Y. N. Bubnov and Y. Z. Voloshin, Dalton Trans., 2013, 13667–16678 RSC; (i) A. V. Dolganov, I. G. Belaya and Y. Z. Voloshin, Electrochim. Acta, 2014, 125, 302–306 CrossRef CAS PubMed.
- (a) Y.-Y. Zhang, Y.-J. Lin and G.-X. Jin, Chem. Commun., 2014, 50, 2327–2329 RSC; (b) M. D. Wise, A. Ruggi, M. Pascu, R. Scopelliti and K. Severin, Chem. Sci., 2013, 4, 1658–1662 RSC.
- M. D. Wise, J. J. Holstein, P. Pattison, C. Besnard, E. Solari, R. Scopelliti, G. Bricogne and K. Severin, Chem. Sci., 2015, 6, 1004–1010 RSC.
- A. M. Sargeson, Coord. Chem. Rev., 1996, 151, 89–114 CrossRef CAS.
- Y. Z. Voloshin, O. A. Varzatskii and Y. N. Bubnov, Russ. Chem. Bull., 2007, 56, 577–605 CrossRef CAS.
- K. Gunasekaran, B. Ma and R. Nussinov, Proteins, 2004, 57, 433–443 CrossRef CAS PubMed.
- Y. Voloshin, O. Varzatskii, S. Shul’ga, V. Novikov, A. Belov, I. Makarenko, I. Dubey, D. Krivorotenko, V. Negrutska, K. Zhizhin, N. Kuznetsov and Y. Bubnov, EUROBIC10, Eur. Biol. Inorg. Chem. Conf., 10th, 2010, 29–38 CAS.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS PubMed.
- R. Bakry, R. M. Vallant, M. Najam-ul-Haq, M. Rainer, Z. Szabo, C. W. Huck and G. K. Bonn, Int. J. Nanomed., 2007, 24, 639–649 Search PubMed.
- (a) R. N. Grimes, Carboranes, Academic Press, 2nd edn, 2011 Search PubMed; (b) Boron Sciences: New technologies and Applications, ed. N. S. Hosmane, CRC Press, 2011 Search PubMed; (c) N. S. Hosmane, J. A. Maguire, Z. Yinghuai and M. Takagaki, Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment, World Scientific Publishers, 2012 Search PubMed; (d) J. Poater, M. Sola, C. Vinas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed; (e) Z. J. Leśnikowski, Collect. Czech. Chem. Commun., 2007, 72, 1646–1658 CrossRef; (f) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701 CrossRef CAS PubMed; (g) M. Scholz, E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 Search PubMed.
- (a) C. E. Wagner, M. L. Mohler, G. S. Kang, D. D. Miller, E. E. Geisert, Y.-A. Chang, E. B. Fleischer and K. J. Shea, J. Med. Chem., 2003, 46, 2823–2833 CrossRef CAS PubMed and references therein; (b) I. Papanastasiou, A. Tsotinis, N. Kolocouris, S. R. Prathalingam and J. M. Kelly, J. Med. Chem., 2008, 51, 1496–1500 CrossRef CAS PubMed and references therein.
- (a) Y. Z. Voloshin, O. A. Varzatskii, A. V. Palchik, I. I. Vorontsov, M. Y. Antipin and E. G. Lebed, Inorg. Chim. Acta, 2005, 358, 131–146 CrossRef CAS PubMed; (b) Y. Z. Voloshin, O. A. Varzatskii, A. V. Palchik, Z. A. Starikova, M. Y. Antipin, E. G. Lebed and Y. N. Bubnov, Inorg. Chim. Acta, 2006, 359, 553–569 CrossRef CAS PubMed; (c) A. S. Belov, A. V. Vologzhanina, V. V. Novikov, V. V. Negrutska, I. Y. Dubey, Z. A. Mikhailova, E. G. Lebed and Y. Z. Voloshin, Inorg. Chim. Acta, 2014, 421, 300–306 CrossRef CAS PubMed.
- (a) Y. Z. Voloshin, V. E. Zavodnik, O. A. Varzatskii, V. K. Belsky, A. V. Palchik, N. G. Strizhakova, I. I. Vorontsov and M. Y. Antipin, J. Chem. Soc., Dalton Trans., 2002, 1193–1202 RSC; (b) Y. Z. Voloshin, O. A. Varzatskii, P. A. Stuzhin, S. V. Shul’ga, S. V. Volkov, A. V. Vologzhanina, E. G. Lebed and Y. N. Bubnov, Inorg. Chem. Commun., 2011, 14, 1504–1507 CrossRef PubMed; (c) Y. Z. Voloshin, O. A. Varzatskii, S. V. Shul’ga, I. N. Denisenko, A. V. Vologzhanina and Y. N. Bubnov, Inorg. Chem. Commun., 2012, 17, 128–131 CrossRef CAS PubMed; (d) O. A. Varzatskii, I. N. Denisenko, S. V. Volkov, A. V. Dolganov, A. V. Vologzhanina, Y. N. Bubnov and Y. Z. Voloshin, Inorg. Chem. Commun., 2013, 33, 147–150 CrossRef CAS PubMed; (e) O. I. Artyushin, I. L. Odinets, E. V. Matveeva, A. V. Vologzhanina, O. A. Varzatskii, S. E. Lubimov and Y. Z. Voloshin, Dalton Trans., 2014, 9677–9689 RSC; (f) O. A. Varzatskii, I. N. Denisenko, A. S. Belov, A. V. Vologzhanina, Y. N. Bubnov, S. V. Volkov and Y. Z. Voloshin, Inorg. Chem. Commun., 2014, 44, 134–138 CrossRef CAS PubMed.
- (a) Y. Z. Voloshin, O. A. Varzatskii, T. E. Kron, V. K. Belsky, V. E. Zavodnik, N. G. Strizhakova and A. V. Palchik, Inorg. Chem., 2000, 39, 1907–1918 CrossRef CAS; (b) Y. Z. Voloshin, O. A. Varzatskii, T. E. Kron, V. K. Belsky, V. E. Zavodnik, N. G. Strizhakova, V. A. Nadtochenko and V. A. Smirnov, J. Chem. Soc., Dalton Trans., 2002, 1203–1211 RSC; (c) Y. Z. Voloshin, O. A. Varzatskii, I. N. Denisenko, S. V. Volkov, A. S. Belov, A. V. Dolganov, A. V. Vologzhanina, V. V. Novikov and Y. N. Bubnov, Eur. J. Inorg. Chem., 2013, 3178–3184 Search PubMed; (d) A. V. Dolganov, A. S. Belov, V. V. Novikov, A. V. Vologzhanina, G. V. Romanenko, Y. G. Budnikova, G. E. Zelinskii, M. I. Buzin and Y. Z. Voloshin, Dalton Trans., 2014, 2476–2487 Search PubMed.
- M. Y. Losytskyy, V. B. Kovalska, O. A. Varzatskii, A. M. Sergeev, S. M. Yarmoluk and Y. Z. Voloshin, J. Fluoresc., 2013, 23, 889–895 CrossRef CAS PubMed.
- G. R. Bardajee, Z. Hooshyar, P. Shafagh, S. Ghiasvand and N. Kakavand, J. Lumin., 2014, 156, 55–62 CrossRef CAS PubMed.
- Z. Hooshyar, G. R. Bardajee, P. Shafagh, S. Ghiasvand, M. Khanjari and N. Dianatnejad, J. Iran. Chem. Soc., 2015, 12, 715–725 CrossRef CAS.
- Z. Hooshyar, G. R. Bardajee, N. Kakavand, M. Khanjari and N. Dianatnejad, Luminescence, 2014, 30, 538–548 CrossRef PubMed.
- Y. Z. Voloshin, O. A. Varzatskii, V. V. Novikov and Y. N. Bubnov, EUROBIC 9, Proc. Eur. Biol. Inorg. Chem. Conf., 9th, 2008, 71–76 CAS.
- (a) V. V. Novikov, O. A. Varzatskii, V. V. Negrutska, Y. N. Bubnov, L. G. Palchykovska, I. Y. Dubey and Y. Z. Voloshin, J. Inorg. Biochem., 2013, 124, 42–45 CrossRef CAS PubMed; (b) Y. Z. Voloshin, V. V. Novikov, O. A. Varzatskii, V. V. Negrutska, L. G. Palchykovska, I. Y. Dubey and Y. N. Bubnov, Patent RU 2012/106191, 2012.
- P. Cigler, M. Kozisek, P. Rezacova, J. Brynda, Z. Otwinowski, J. Pokorna, J. Plesek, B. Gruner, L. Doleckova-Maresova, M. Masa, J. Sedlacek, J. Bodem, H. G. Krausslich, V. Kral and J. Konvalinka, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15394–15399 CrossRef CAS PubMed.
- M. Kozisek, P. Cigler, M. Lepsik, J. Fanfrlik, P. Rezacova, J. Brynda, J. Pokorna, J. Plesek, B. Gruner, K. Grantz Saskova, J. Vaclavikova, V. Kral and J. Konvalinka, J. Med. Chem., 2008, 51, 4839–4843 CrossRef CAS PubMed.
- (a) O. A. Varzatskii, V. V. Novikov, S. V. Shulga, A. S. Belov, A. V. Vologzhanina, V. V. Negrutska, I. Y. Dubey, Y. N. Bubnov and Y. Z. Voloshin, Chem. Commun., 2014, 50, 3166–3168 RSC; (b) O. A. Varzatskii, S. V. Shul’ga, A. S. Belov, V. V. Novikov, A. V. Dolganov, A. V. Vologzhanina and Y. Z. Voloshin, Dalton Trans., 2014, 17934–17948 RSC.
- V. B. Kovalska, M. Y. Losytskyy, O. A. Varzatskii, V. V. Cherepanov, Y. Z. Voloshin, A. A. Mokhir, S. M. Yarmoluk and S. V. Volkov, Bioorg. Med. Chem., 2014, 22, 1883–1888 CrossRef CAS PubMed.
- H. Deng and V. A. Bloomfield, Biophys. J., 1999, 77, 1556–1561 CrossRef CAS.
- (a) D. L. Bailey, D. W. Townsend, P. E. Valk and M. N. Maisey, Positron Emission Tomography: Basic Sciences, Springer, Secaucus, NJ, 2005 Search PubMed; (b) P. S. Donnelly, Dalton Trans., 2011, 999–1010 RSC; (c) V. Amendola, Coord. Chem. Rev., 2006, 250, 1451–1470 CrossRef CAS PubMed.
- P. J. Blower, J. S. Lewis and J. Zweit, Nucl. Med. Biol., 1996, 23, 957–980 CrossRef CAS.
- (a) S. V. Smith, J. Inorg. Biochem., 2004, 98, 1874–1901 CrossRef CAS PubMed; (b) N. D. Bartolo, A. M. Sargeson and S. V. Smith, Org. Biomol. Chem., 2006, 4, 3350–3357 RSC; (c) S. D. Voss, S. V. Smith, N. D. Bartolo, L. J. McIntosh, E. M. Cyr, A. A. Bonab, E. A. Carter, A. J. Fischman, S. T. Treves, S. D. Gillies, A. M. Sargeson, J. S. Huston and A. B. Packard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17489–17493 CrossRef CAS PubMed.
- H. Cai, J. Fissekis and P. S. Conti, Dalton Trans., 2009, 5395–5400 RSC.
- H. Cai, Z. Li, C.-W. Huang, R. Park, A. H. Shahinian and P. S. Conti, Nucl. Med. Biol., 2010, 37, 57–65 CrossRef CAS PubMed.
- S. Liu, Z. Li, L.-P. Yap, C.-W. Huang, R. Park and P. S. Conti, Chem.–Eur. J., 2011, 17, 10222–10225 CrossRef CAS PubMed.
- M. T. Ma, O. C. Neels, D. Denoyer, P. Roselt, J. A. Karas, D. B. Scanlon, J. M. White, R. J. Hicks and P. S. Donnelly, Bioconjugate Chem., 2011, 22, 2093–2103 CrossRef CAS PubMed.
- M. T. Ma, J. A. Karas, J. M. White, D. Scanlon and P. S. Donnelly, Chem. Commun., 2009, 3237–3239 RSC.
- M. T. Ma, M. S. Cooper, R. L. Paul, K. P. Shaw, J. A. Karas, D. Scanlon, J. M. White, P. J. Blower and P. S. Donnelly, Inorg. Chem., 2011, 50, 6701–6710 CrossRef CAS PubMed.
- K. Chen, X. Wang, W.-Y. Lin, C. K.-F. Shen, L.-P. Yap, L. D. Hughes and P. S. Conti, ACS Med. Chem. Lett., 2012, 3, 1019–1023 CrossRef CAS PubMed.
- B. M. Paterson, G. Buncic, L. E. McInnes, P. Roselt, C. Cullinane, D. S. Binns, C. M. Jeffery, R. I. Price, R. J. Hicks and P. S. Donnelly, Dalton Trans., 2015, 4901–4909 RSC.
- S. Liu, Z. Li and P. S. Conti, Molecules, 2014, 19, 4246–4255 CrossRef PubMed.
- G. Li, X. Wang, S. Zong, J. Wang, P. S. Conti and K. Chen, Mol. Pharm., 2014, 11, 3938–3946 CrossRef CAS PubMed.
- Z. Wu, S. Liu, I. Nair, K. Omori, S. Scott, I. Todorov, J. E. Shively, P. S. Conti, Z. Li and F. Kandeel, Theranostics, 2014, 4, 770–777 CrossRef CAS PubMed.
- C. Brand, D. Abdel-Atti, Y. Zhang, S. Carlin, S. M. Clardy, E. J. Keliher, W. A. Weber, J. S. Lewis and T. Reiner, Bioconjugate Chem., 2014, 25, 1323–1330 CrossRef CAS PubMed.
- S. Liu, D. Li, C.-W. Huang, L.-P. Yap, R. Park, H. Shan, Z. Li and P. S. Conti, Mol. Imag. Biol., 2012, 14, 718–724 CrossRef PubMed.
- S. Liu, D. Li, C.-W. Huang, L.-P. Yap, R. Park, H. Shan, Z. Li and P. S. Conti, Theranostics, 2012, 2, 589–596 CrossRef CAS PubMed.
- K. V. Tan, P. A. Pellegrini, B. W. Skelton, C. F. Hogan, I. Greguric and P. J. Barnard, Inorg. Chem., 2014, 53, 468–477 CrossRef CAS PubMed.
- B. M. Paterson, P. Roselt, D. Denoyer, C. Cullinane, D. Binns, W. Noonan, C. M. Jeffery, R. I. Price, J. M. White, R. J. Hicks and P. S. Donnelly, Dalton Trans., 2014, 1386–1396 RSC.
- S. V. Smith, J. M. Harrowfield, N. M. Di Bartolo and A. M. Sargeson, PCT Int. Appl. WO 00/40585, 2000.
- K. Alt, B. M. Paterson, K. Ardipradja, C. Schieber, G. Buncic, B. Lim, S. S. Poniger, B. Jakoby, X. Wang, G. J. O’Keefe, H. J. Tochon-Danguy, A. M. Scott, U. Ackermann, K. Peter, P. S. Donnelly and C. E. Hagemeyer, Mol. Pharm., 2014, 11, 2855–2863 CrossRef CAS PubMed.
- B. M. Paterson, K. Alt, C. M. Jeffery, R. I. Price, S. Jagdale, S. Rigby, C. C. Williams, K. Peter, C. E. Hagemeyer and P. S. Donnelly, Angew. Chem., Int. Ed., 2014, 53, 6115–6119 CrossRef CAS PubMed.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS PubMed.
- C.-W. Huang, Z. Li, H. Cai, K. Chen, T. Shahinian and P. S. Conti, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 1313–1322 CrossRef CAS PubMed.
- S. M. Janib, S. Liu, R. Park, M. K. Pastuszka, P. Shi, A. S. Moses, M. M. Orosco, Y.-A. Lin, H. Cui and P. S. Conti, Integr. Biol., 2013, 5, 183–194 RSC.
- I. Bertini, C. Luchinat and G. Parigi, Solution NMR of paramagnetic molecules: applications to metallobiomolecules and models, Elsevier, 2001, vol. 2 Search PubMed.
- M. A. Hass and M. Ubbink, Curr. Opin. Struct. Biol., 2014, 24, 45–53 CrossRef CAS PubMed.
- C. Luchinat, G. Parigi, E. Ravera and M. Rinaldelli, J. Am. Chem. Soc., 2012, 134, 5006–5009 CrossRef CAS PubMed.
- J. Iwahara and G. M. Clore, Nature, 2006, 440, 1227–1230 CrossRef CAS PubMed.
- T. Grobner and F. C. Prischl, Kidney Int., 2007, 72, 260–264 CrossRef CAS PubMed.
- D. E. Sosnovik and P. Caravan, Current Cardiovascular Imaging Reports, 2013, 6, 61–68 CrossRef PubMed.
- R. D. Bolskar, Nanomedicine, 2008, 3, 201–213 CrossRef CAS PubMed.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- É. Tóth, R. D. Bolskar, A. Borel, G. González, L. Helm, A. E. Merbach, B. Sitharaman and L. J. Wilson, J. Am. Chem. Soc., 2005, 127, 799–805 CrossRef PubMed.
- (a) S. Laus, B. Sitharaman, É. Tóth, R. D. Bolskar, L. Helm, L. J. Wilson and A. E. Merbach, J. Phys. Chem. C, 2007, 111, 5633–5639 CrossRef CAS; (b) S. Laus, B. Sitharaman, É. Tóth, R. D. Bolskar, L. Helm, S. Asokan, M. S. Wong, L. J. Wilson and A. E. Merbach, J. Am. Chem. Soc., 2005, 127, 9368–9369 CrossRef CAS PubMed.
- C. Felton, A. Karmakar, Y. Gartia, P. Ramidi, A. S. Biris and A. Ghosh, Drug Metab. Rev., 2014, 46, 142–154 CrossRef CAS PubMed.
- (a) R. D. Bolskar, Nanomedicine, 2008, 3, 201–221 CrossRef CAS PubMed; (b) P. P. Fatouros and M. D. Shultz, Nanomedicine, 2013, 8, 1853–1864 CrossRef CAS PubMed.
- L. S. Szczepaniak, A. Sargeson, I. I. Creasei, R. J. Geue, M. Tweedle and R. G. Bryant, Bioconjugate Chem., 1992, 3, 27–31 CrossRef CAS.
- W.-M. Liu, M. Overhand and M. Ubbink, Coord. Chem. Rev., 2014, 273–274, 2–12 CrossRef CAS PubMed.
- G. Otting, Annu. Rev. Biophys., 2010, 39, 387–405 CrossRef CAS PubMed.
- G. Pintacuda, A. Moshref, A. Leonchiks, A. Sharipo and G. Otting, J. Biomol. NMR, 2004, 29, 351–361 CrossRef CAS.
- M. Prudencio, J. Rohovec, J. A. Peters, E. Tocheva, M. J. Boulanger, M. E. P. Murphy, H. J. Hupkes, W. Kosters, A. Impagliazzo and M. Ubbink, Chem.–Eur. J., 2004, 10, 3252–3260 CrossRef CAS PubMed.
- P. H. J. Keizers, J. F. Desreux, M. Overhand and M. Ubbink, J. Am. Chem. Soc., 2007, 129, 9292–9293 CrossRef CAS PubMed.
- A. A. Bothner-By, P. J. Domaille and C. Gayathri, J. Am. Chem. Soc., 1981, 103, 5602–5603 CrossRef CAS.
- M. Piccioli and P. Turano, Coord. Chem. Rev., 2015, 284, 313–328 CrossRef CAS PubMed.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- Y. Z. Voloshin, A. Y. Lebedev, V. V. Novikov, A. V. Dolganov, A. V. Vologzhanina, E. G. Lebed, A. A. Pavlov, Z. A. Starikova, M. I. Buzin and Y. N. Bubnov, Inorg. Chim. Acta, 2013, 399, 67–78 CrossRef CAS PubMed.
- Y. Z. Voloshin, O. A. Varzatskii, V. V. Novikov, N. G. Strizhakova, I. I. Vorontsov, A. V. Vologzhanina, K. A. Lyssenko, G. V. Romanenko, M. V. Fedin, V. I. Ovcharenko and Y. N. Bubnov, Eur. J. Inorg. Chem., 2010, 5401–5415 CrossRef CAS PubMed.
- O. A. Varzatskii, L. V. Penkova, S. V. Kats, A. V. Dolganov, A. V. Vologzhanina, A. A. Pavlov, V. V. Novikov, A. S. Bogomyakov, V. N. Nemykin and Y. Z. Voloshin, Inorg. Chem., 2014, 53, 3062–3071 CrossRef CAS PubMed.
- V. V. Novikov, A. A. Pavlov, Y. V. Nelyubina, M.-E. Boulon, O. A. Varzatskii, Y. Z. Voloshin and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137, 9792–9795 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |