
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-catalyzed C–N coupling of vinylbromides and sulfonimidamides: a facile synthesis of N′-vinylsulfonimidamides†
Ganesh C.
Nandi
ab,
Sudhakar R.
Kota
a,
Prasad B.
Wakchaure
c,
Praveen K.
Chinthakindi
a,
Thavendran
Govender
a,
Hendrick G.
Kruger
a,
Tricia
Naicker
*a and
Per I.
Arvidsson
*ad
aCatalysis and Peptide Research Unit, University of KwaZulu-Natal, Durban, South Africa. E-mail: Per.Arvidsson@scilifelab.se
bOrganic Chemistry Section, National Institute for Interdisciplinary Science and Technology (CSIR), Trivandrum 695019, India
cOrganic Pharmaceutical Chemistry, Department of Medicinal Chemistry, Uppsala Biomedical Center, Uppsala University, P.O. Box 574, 751 23 Uppsala, Sweden
dScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden
First published on 20th July 2015
Abstract
N′-Vinyl sulfonimidamides have been synthesized through a Pd-catalyzed C–N cross coupling between the N′-(imine nitrogen) of N′-deprotected sulfonimidamides and vinyl bromides. The hitherto unreported products were obtained in moderate to excellent yield, and the C–C double bond geometry of the vinylic substrates were retained during the course of reaction. Single crystal X-ray crystallographic analysis confirmed the product structure. Furthermore, we demonstrate that the formed N′-vinyl sulfonimidamides could undergo hydrogenation with Pd–C/H2 to provide N′-alkyl sulfonimidamides.
Introduction
As part of our ongoing research project on sulfonimidamides and acyl sulfonimidamides as bioisosteres in medicinal chemistry,1 we recently disclosed new synthetic protocols for preparation and modification of this functional group. In our first report,2a we used a Pd-catalyzed Buchwald-Hartwig2f,g C–N coupling procedure for the arylation of the N′-(imine nitrogen) of sulfonimidamides. Subsequently, we reported the synthesis of N-acylated sulfonimidamides via Pd-catalyzed amidocarbo-nylation of vinyl/aryl halides and triflates.2b,c We have also reported N′-arylation via Cu-mediated Chan–Lam–Evans coupling,2d and N-alkynylation through a Cu-catalyzed dual N–H/C–H activation protocol.2e Herein, we explore the Pd-catalyzed C–N coupling of various vinyl bromides to the N′-(imine nitrogen) of unprotected sulfonimidamides.Although Levchenko et al.,3 reported the synthesis of sulfonimidamides already in 1960, the interest in this functional group has remained low until quite recently. However, during the last decade, attention on this functional group has increased tremendously, both in the area of synthetic methodology and for applications in medicinal chemistry and agrochemistry. Some research groups applied sulfonimidamides as a reagent in organic synthesis, mainly as a ‘N’-source for metal-catalyzed nitrene transfer reactions, for imination of sulfides, aziridination of olefins, and C–H aminations of hydrocarbons.4 Additionally, sulfonimidamides have been used as organocatalysts and as chiral ligands in asymmetric synthesis; more recently they have been used in the iridium-catalyzed asymmetric hydrogenation of cyclic enamides.5 Sulfonimidamide functional groups have also been used as analogues of oncolytic sulfonylureas, sodium channel antagonists, pesticidal agents, and as transition state analogue inhibitors of aspartic acid metalloproteases.6 Even more recently, other groups, in addition to ours, have reported the synthetic methodology for sulfonimidamide functionalization and preparation.7 Given the growing bio- and synthetic importance of sulfonimidamides, we became interested in attempting to prepare N-vinyl sulfonimidamides following the Pd-catalyzed Buchwald–Hartwig8 C–N coupling procedure. To the best of our knowledge, there is no report on the synthesis of N-vinyl sulfonimidamides through any methodology.
The N-alkenyl functional group represent a very versatile class of olefinic compounds as the electron releasing ability of the nitrogen lone pair strongly polarizes the double bond; thereby offering high levels of reactivity in combination with strong differentiation of the two sp2 carbon atoms. In fact, this reactivity of N-alkenyls is the basis for enamine activation in the very fruitful research area of organocatalysis9–11 – another area of interest in our research group. Consequently, N-alkenyl functional groups are widely used as synthetic intermediates in the preparation of heterocycles12 and in asymmetric synthesis of amides and amino acids.13 In addition, N-alkenyl functional groups can be found in many natural products, that include anti-anthelmintic chondriamides,14 cytotoxic tripeptide caspergillamides,15 protease inhibitors TMC-95-A-D,16 antibiotic CJ-15801,17 anti-inflammatory frangufoline,18 as well as antitumor lobatomide A–F,19 salicylihalamide A and related compounds.20
Results and discussion
The starting sulfonimidamides were not commercially accessible, and hence prepared according to literature procedures (Scheme 1).2a,21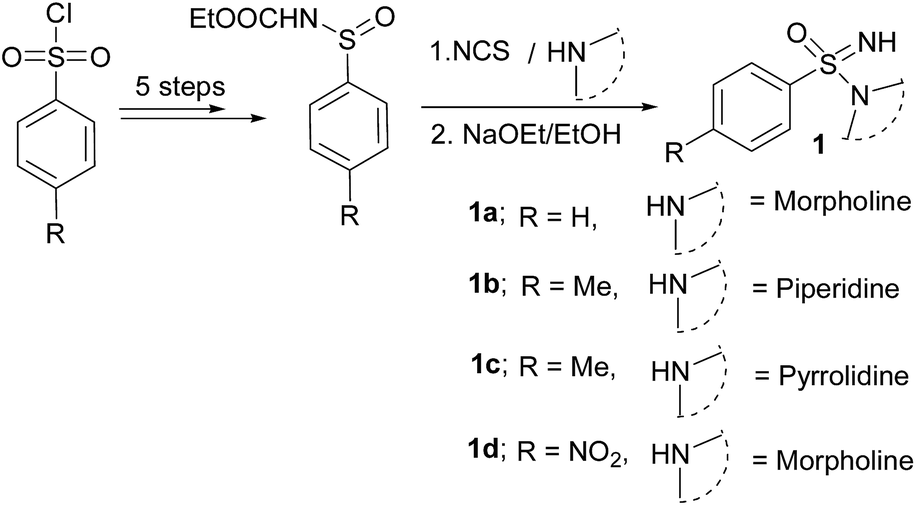 | ||
| Scheme 1 Synthesis of sulfonimidamides. | ||
We commenced our studies with the reaction of sulfonimidamide 1a and 1-(1-bromovinyl)-4-chlorobenzene as model substrates for optimization and the results have been depicted in Table 1. First, we attempted our previously reported reaction condition2a (Table 1, entry 1) for N-arylation of sulfonimidamide [i.e. catalyst Pd(PhCH2CH2NH2)(Cl) (RuPhos) and NaOtBu base in THF under MW irradiation at 100 °C]; surprisingly, this did not lead to any detectable N′-vinylated product. Next, we attempted the model reaction with the catalyst Pd2(dba)3 (1 mol%), ligand BINAP (1.5 mol%), base NaOtBu (1.5 equiv.) in toluene at 100 °C for 12 h; gratifyingly, these reaction conditions gave 78% of the expected product with some unreacted starting materials (Table 1, entry 2). A longer reaction time did not improve the yield of the reaction, but performing the reaction at 110 °C, provided 92% of isolated product within 4 h (Table 1, entry 3). Pd(OAc)2 was found to be similarly effective as Pd2(dba)3 and afforded 89% of the product (Table 1, entry 4). Interestingly, under MW irradiation the reaction completed within 1.5 h but provided only 82% of product (Table 1, entry 5). We were also able to demonstrate that the reaction could be performed under another Buchwald's procedure22 for N-vinylation, i.e., stoichiometric CuI/N,N′-dimethylethylenediamine (DMEDA) mediation, rather than catalytic Pd-/BINAP conditions, albeit with longer reactions times and lower yield (79%) of product (Table 1, entry 6).
|
|
||
|---|---|---|
| Entry | Condition | Yieldb (%) |
| a Sulfonimidamide and vinyl bromide were used 1.0 equiv. each. b Isolated yield. c Some starting materials were unreacted. | ||
| 1 | Pd(PhCH2CH2NH2)(Cl)(RuPhos) (3.0 mol%), NaOtBu (1.2 equiv.), THF, 100 °C, MW, 2.5 h | — |
| 2 | Pd2(dba)3 (1 mol%), BINAP (1.5 mol%), NaOtBu (1.5 equiv.), toluene, 100 °C, 12 h | 78c |
| 3 | Pd2(dba)3 (1 mol%), BINAP (1.5 mol%), NaOtBu (1.5 equiv.), toluene, 110 °C, 4 h | 92 |
| 4 | Pd(OAc)2 (1 mol%), BINAP (1.5 mol%), NaOtBu (1.5 equiv.), toluene, 110 °C, 5 h | 89 |
| 5 | Pd2(dba)3 (1 mol%), BINAP (1.5 mol%), NaOtBu (1.5 equiv.), toluene, 150 °C, MW, 1.5 h | 82 |
| 6 | CuI (1.0 equiv.), DMEDA (2.0 equiv.), K2CO3(2.0 equiv.), toluene, 110 °C, 7 h | 79 |
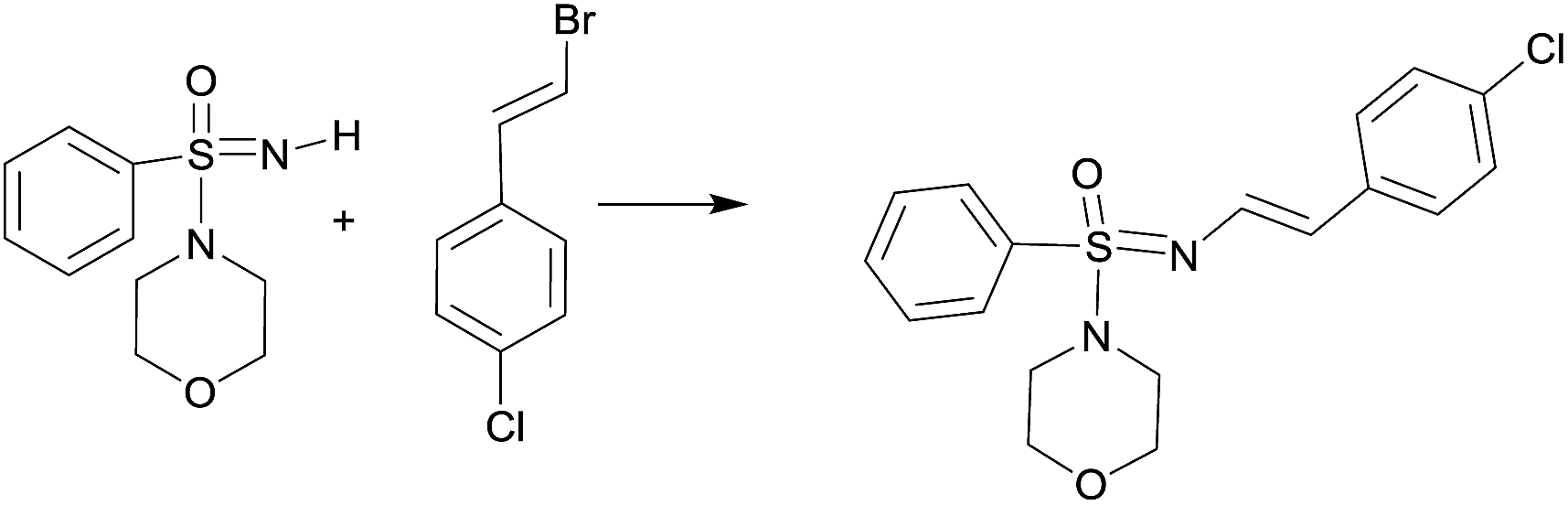
With the optimized reaction conditions (Table 1, entry 3) at hand; we next investigated the substrate scope of this N′-vinylation reaction by varying vinyl halides and sulfonimidamides. Representative results are shown in Chart 1. Various aryl vinylbromides were successfully employed for this transformation. Both unsubstituted- and –Cl/–OMe substituted aryl (trans-) vinyl bromides provided the corresponding N′-vinyl sulfonimidamides (3a–c, 3h–j, 3o, p) in good to excellent yields. The phenylvinyl bromide with cis-orientation also gave satisfactory results whilst retaining the double-bond geometry (3e, 3l, 3n). Vinylbromides with heteroaryl groups were equally effective for this transformation (3d, 3k).
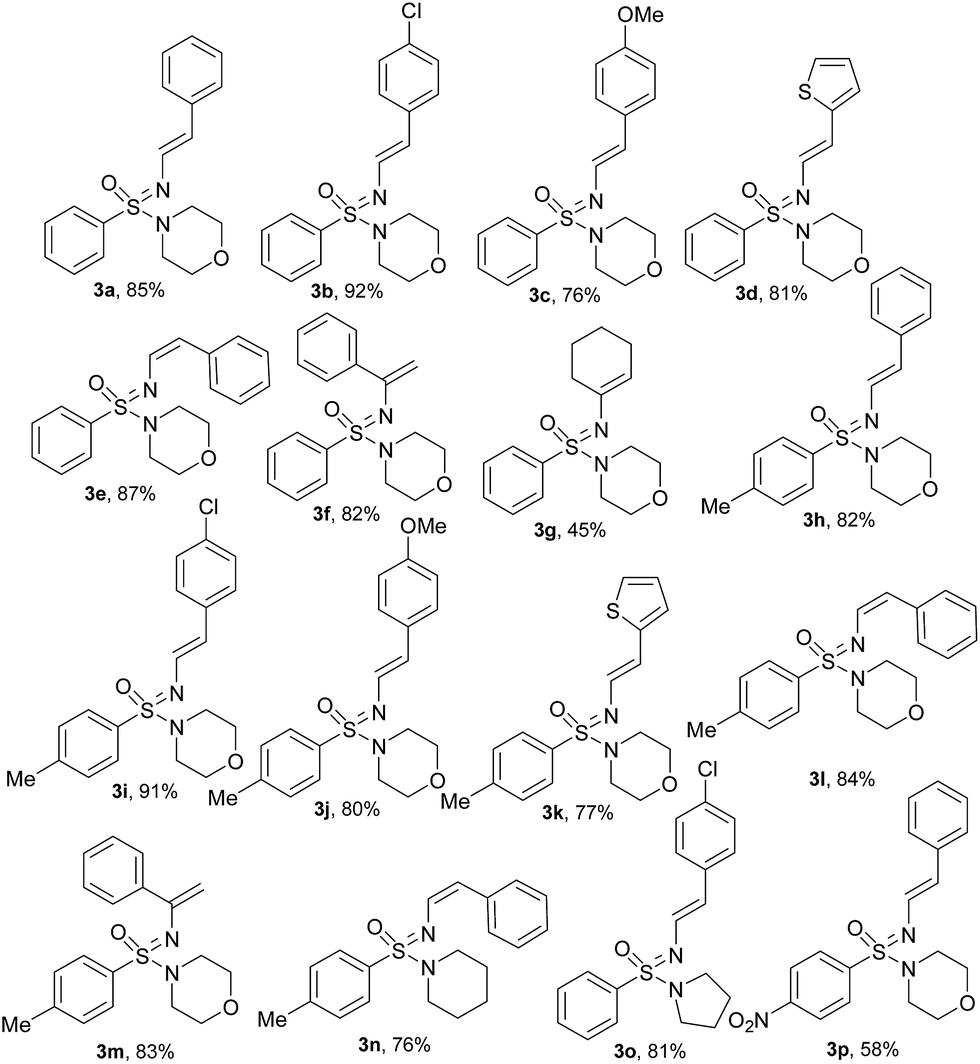 | ||
| Chart 1 Substrate scope for the N′-vinylation of the sulfonimidamides under optimized reaction conditions. | ||
Although the cyclic aliphatic vinyl bromide afforded the corresponding product (3g) in moderate yield, both acyclic aliphatic vinyl bromides 2-bromo-1-propene and 1-bromo-1-propene resulted a complex reaction mixture. Even α-bromo styrene worked well for this transformation and provided very good yields of terminal alkenes (3f, 3m). Modification of the sulfonimidamide moiety was tolerated satisfactorily as shown by replacing the morpholine functionality with piperidine/pyrrolidine to offer the products (3n, 3o) in very good yields.
The structures of all the newly synthesized compounds were deduced from full spectroscopic characterization and unequivocally established by X-ray single crystal diffraction analysis of one representative compound (3c, Fig. 1).23 According to a search performed using the Cambridge Structural Database, this structure is the first of its kind to be reported. Comparable sulfonimidamide and sulfonimidate structures displayed a fairly trigonal pyramidal shape with respect to the groups bonded to sulphur atom; bond angles S1–O3–C13 109°, S1–O3–N1 104°, while S1–O3–N2 122° (deviation). In addition, the X-ray structure of 3c displayed a slightly longer S1–N1 bond length of 1.6 Å as compared to analogous structures that displayed approximately 1.5 Å. The S1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N2 double bond length in the structure of 3c is 1.51 Å, which is also slightly longer than other reported structures (1.4–1.52 Å). Similar to other enamine systems, the CC bond displays (E)-configuration, and the conformation of the bond between the N′-atom and the C3 is trans-, so that the styryl group points away from the bulky substituents of the sulfone group and the conjugated π-system that includes the N′-atom, the CC bond, and the phenyl ring, is perfectly planar.
N2 double bond length in the structure of 3c is 1.51 Å, which is also slightly longer than other reported structures (1.4–1.52 Å). Similar to other enamine systems, the CC bond displays (E)-configuration, and the conformation of the bond between the N′-atom and the C3 is trans-, so that the styryl group points away from the bulky substituents of the sulfone group and the conjugated π-system that includes the N′-atom, the CC bond, and the phenyl ring, is perfectly planar.
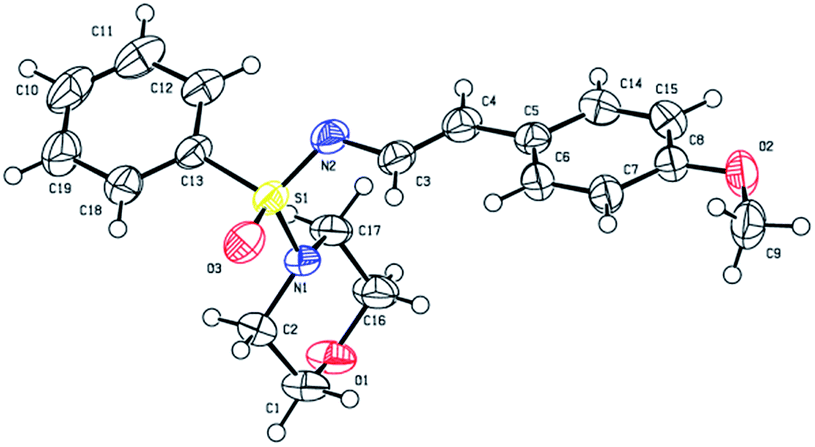 | ||
| Fig. 1 ORTEP diagram of 3c. | ||
The N′-alkenyl compounds reported here are expected to be very sensitive towards acid. Not too surprising, the N-vinyl sulfonimidamides 3 decomposed to starting sulfonimidamide 4 and aryl acetaldehyde 5 (Scheme 2) when CDCl3 was used as solvent for recording the NMR spectra. Hence, we replaced CDCl3 by CD3CN as solvent for NMR spectroscopic characterization of the products. For authentication, in a separate experiment, sulfonimidamide 4 and phenyl acetaldehyde (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were mixed in CDCl3 in an NMR tube; the resulting NMR spectra matched that obtained from the reaction product 3a in CDCl3, thus independently verifying the decomposition of 3 to 4 and 5 under slightly acidic conditions. To verify the reversibility of the process, sulfonimidamide 4 and phenyl acetaldehyde 5 (1:1) were refluxed in toluene in the presence of catalytic p-toluenesulfonic acid (PTSA); LCMS analysis of the reaction mixture confirmed the formation of 3 albeit in trace amount.
:1) were mixed in CDCl3 in an NMR tube; the resulting NMR spectra matched that obtained from the reaction product 3a in CDCl3, thus independently verifying the decomposition of 3 to 4 and 5 under slightly acidic conditions. To verify the reversibility of the process, sulfonimidamide 4 and phenyl acetaldehyde 5 (1:1) were refluxed in toluene in the presence of catalytic p-toluenesulfonic acid (PTSA); LCMS analysis of the reaction mixture confirmed the formation of 3 albeit in trace amount.
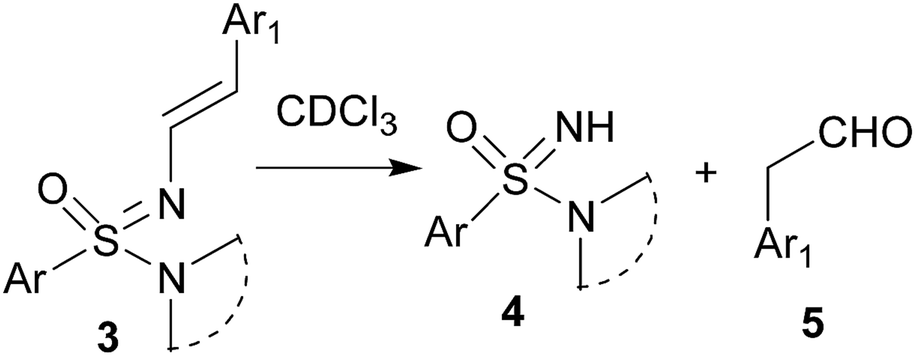 | ||
| Scheme 2 Decomposition of N′-vinyl sulfonimidamides in CDCl3. | ||
In order to demonstrate the reactivity and utility of the newly prepared N-vinyl sulfonimidamides, we decided to reduce the N-alkenyl group to the corresponding N-alkane derivative. An attempt to reduce the double bond following our previously reported BH3·DMS mediated reduction2e proved unsuccessful. However, for these substrates Pd–C catalyzed hydrogenation provided the expected N′-alkyl sulfonimidamides 6 in good yield (Scheme 3). Under the same conditions, compound 3p gave the corresponding amine 6b through a one-pot reduction of both the olefin and the nitro functionality, thus making an interesting building block for further transformations.
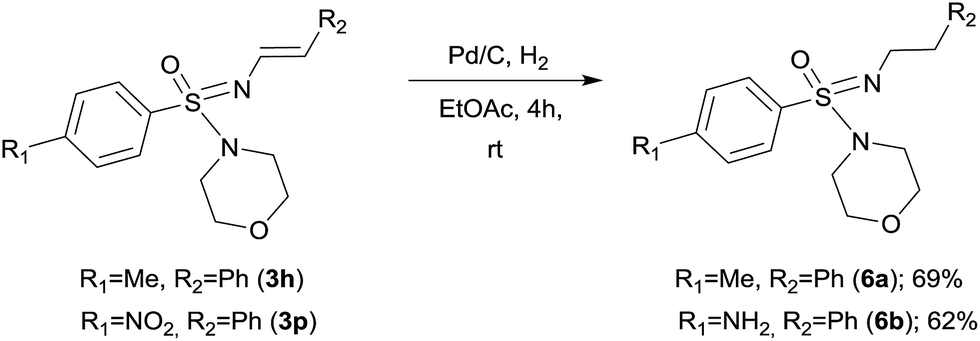 | ||
| Scheme 3 Pd–C catalyzed hydrogenation of N′-vinyl sulfonimidamides. | ||
Conclusions
In summary, we have described a Pd-catalyzed C–N coupling reaction of N′-deprotected sulfonimidamides and vinyl bromides for the synthesis of N′-vinyl sulfonimidamides. A variety of α/β-vinyl bromides and cyclic vinyl bromides were successfully coupled to different N′-deprotected sulfonimidamides. The coupling took place with retained stereochemistry around the vinylic double bond. The structure of the N′-vinyl sulfonimidamide product was indisputably proven by single crystal X-ray diffraction. In addition, we demonstrated the reactivity of the newly formed N′-alkenyl group through hydrogenation of N′-vinyl sulfonimidamides to N′-alkyl sulfonimidamides. The preliminary observation of the dynamic nature of these N-vinyl sulfonimidamides suggest that they can be used as precursors for many other synthetic transformations and further studies along these lines are currently underway in our laboratory.Experimental
General
The starting material sulfonimidamides were synthesized in the laboratory following the reported method.2a,21 Vinyl bromide [(E)-2-bromoethenyl]benzene and 1-[(E)-2-bromoethenyl]-4-methoxybenzene,24 1-[(E)-2-bromoethenyl]-4-chlorobenzene,25 2-[(E)-2-bromoethenyl]thiophene,26 [(Z)-2-bromoethenyl]benzene,27 1-bromocyclohex-1-ene28 were synthesized using reported procedures. (1-Bromoethenyl)benzene and Pd-catalysts, ligands were purchased from various suppliers and used as received. 1H and 13C NMR spectra were recorded on Bruker Avance III spectrometer operating at 400 and 100 MHz, respectively. CD3CN and CDCl3 were purchased from Merck and used as received. IR spectra were recorded on BRUKER-ALPHA spectrophotometer. HRMS were recorded on BRUKER micrOTOF-Q-II spectrometer. Melting points were uncorrected.Acknowledgements
This work is based on research supported in part by the National Research Foundation of South Africa (grant number 87706).Notes and references
- (a) F. Sehgelmeble, J. Janson, C. Ray, S. Rosqvist, S. Gustavsson, L. I. Nilsson, A. Minidis, J. Holenz, D. Rotticci, J. Lundkvist and P. I. Arvidsson, ChemMedChem, 2012, 7, 396 CrossRef CAS PubMed; (b) S. R. Borhade, R. Svensson, P. Brandt, P. Artursson, P. I. Arvidsson and A. Sandstrom, ChemMedChem, 2015, 10, 455 CrossRef CAS PubMed.
- (a) M. F. Maldonado, F. Sehgelmeble, F. Bjarnemark, M. Svensson, J. Ahman and P. I. Arvidsson, Tetrahedron, 2012, 68, 7456 CrossRef; (b) S. R. Borhade, A. Sandstrom and P. I. Arvidsson, Org. Lett., 2013, 15, 1056 CrossRef CAS PubMed; (c) P. B. Wakchaure, S. R. Borhade, A. Sandstrom and P. I. Arvidsson, Eur. J. Org. Chem., 2015, 213 CrossRef CAS; (d) G. C. Nandi, S. R. Kota, T. Govender, H. G. Kruger and P. I. Arvidsson, Tetrahedron, 2014, 70, 5428 CrossRef CAS; (e) G. C. Nandi, S. R. Kota, T. Naicker, T. Govender, H. G. Kruger and P. I. Arvidsson, Eur. J. Org. Chem., 2015, 2861 CrossRef CAS; (f) M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6686 CrossRef CAS PubMed; (g) D. Maiti, B. Fors, J. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011, 2, 57 RSC.
- (a) E. S. Levchenko, I. E. Sheinkman and A. V. Kirsanov, Zh. Obshch. Khim., 1960, 30, 1941 CAS; (b) E. S. Levchenko, N. Y. Derkach and A. V. Kirsanov, Zh. Obshch. Khim., 1962, 32, 1208 CAS.
- (a) D. Leca, A. Toussaint, C. Mareau, L. Fensterbank, E. Lacote and M. Malacria, Org. Lett., 2004, 6, 3573 CrossRef CAS PubMed; (b) P. H. Di Chenna, F. Robert-Peillard, P. Dauban and R. H. Dodd, Org. Lett., 2004, 6, 4503 CrossRef CAS PubMed; (c) C. Liang, F. Robert-Peillard, C. Fruit, P. Muller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641 CrossRef CAS PubMed; (d) C. Fruit, F. Robert-Peillard, G. Bernardinelli, P. Muller, R. H. Dodd and P. Dauban, Tetrahedron: Asymmetry, 2005, 16, 3484 CrossRef CAS; (e) F. Robert-Peillard, P. H. Di Chenna, C. Liang, C. Lescot, F. Collet, R. H. Dodd and P. Dauban, Tetrahedron: Asymmetry, 2010, 21, 1447 CrossRef CAS; (f) F. Collet, R. H. Dodd and P. Dauban, Org. Lett., 2008, 10, 5473 CrossRef CAS PubMed; (g) C. Liang, F. Collet, F. Robert-Peillard, P. Muller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343 CrossRef CAS PubMed; (h) C. Lescot, B. Darses, F. Collet, P. Retailleau and P. Dauban, J. Org. Chem., 2012, 77, 7232 CrossRef CAS PubMed; (i) B. Darses, A. G. Jarvis, A. K.- Mafroud, G. Estenne-Bouhtou, G. Dargazanli and P. Dauban, Synthesis, 2013, 45, 2079 CrossRef CAS; (j) A. Beltran, C. Lescot, M. M. Díaz-Requejo, P. J. Perez and P. Dauban, Tetrahedron, 2013, 69, 4488 CrossRef CAS.
- (a) C. Worch, I. Atodiresei, G. Rabbe and C. Bolm, Chem.–Eur. J., 2010, 16, 677 CrossRef CAS PubMed; (b) C. Worch and C. Bolm, Synlett, 2009, 2425 CAS; (c) M. Steurer and C. Bolm, J. Org. Chem., 2010, 75, 3301 CrossRef CAS PubMed; (d) F. W. Patureau, C. Worch, M. A. Siegler, A. L. Spek, C. Bolm and J. N. H. Reek, Adv. Synth. Catal., 2012, 354, 59 CrossRef CAS.
- (a) J. E. Toth, G. B. Grindey, W. J. Ehlhardt, J. E. Ray, G. B. Boder, J. R. Bewley, K. K. Klinggerman, S. B. Gates, S. M. Rinzel, R. M. Schultz, L. C. Weir and J. F. Worzalla, J. Med. Chem., 1997, 40, 1018 CrossRef CAS PubMed; (b) C. Gnamm, A. Jeanguenat, A. C. Dutton, C. Grimm, D. P. Kloer and J. Crossthwaite, Bioorg. Med. Chem. Lett., 2012, 22, 3800 CrossRef CAS PubMed; (c) H.-W. Kleemann, J. Brendel, J. R. Schwark, A. Weichert, H. J. Lang, U. Albus and W. Scholz, EP0771788 A2, 1997, Hoechst AG, Frankfurt am Main, Germany; (d) H.-W. Kleemann, H. J. Lang, J. R. Schwark, A. Weichert, W. Scholz and U. Albus, US6057322 A, 2000, Hoechst AG, Frankfurt am Main, Germany; (e) R. Paulini, D. Breuninger, D. von Deyn, H. M. M. Bastiaans, C. Beyer, D. D. Anspaugh and H. Oloumi-Sadeghi, WO 2009/156336 A1, BASF SE, Ludwigshafen, Germany, 2009; (f) R. Paulini, D. Breuninger, D. von Deyn, H. M. M. Bastiaans, C. Beyer, WO 2011/069955, BASF SE, Ludwigshafen, Germany, 2011, A1.
- Y. Chen and J. Gibson, RSC Adv., 2015, 5, 4171 RSC.
- Review: (a) A. R. Muci and S. L. Buchwald, Top. Curr.Chem., 2002, 219, 131 CrossRef CAS; (b) J. F. Hartwig, in Modern Amination Methods, ed. A. Ricci, Wiley-VCH, Weinheim, Germany, 2000, pp. 195–262 Search PubMed.
- (a) The Chemistry of Enamines, ed. Z. Rappaport, Wiley, New York, 1994 Search PubMed; (b) Enamines, ed. A. G. Cook, Marcel Decker, New York, 1988 Search PubMed.
- For reviews on enamides, see: (a) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455 RSC; (b) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS PubMed; (c) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed.
- H. Liu, X. Yan, C. Chen, Q. Liu and C. Xi, Chem. Commun., 2013, 49, 5513 RSC.
- (a) A. Furstner, T. Dierkes, O. R. Thiel and G. Blanda, Chem.–Eur. J., 2001, 7, 5286 CrossRef CAS PubMed; (b) A. G. Schultz, P. R. Guzzo and D. M. Nowak, Int. J. Mod. Org. Chem., 1995, 60, 8044 CrossRef CAS; (c) M. A. Brodney and A. Padwa, J. Org. Chem., 1999, 64, 556 CrossRef CAS; (d) D. T. Davies, N. Kapur and A. F. Parsons, Tetrahedron, 2000, 56, 3941 CrossRef CAS.
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) J.-A. Ma, Angew. Chem., Int. Ed., 2003, 42, 4290 CrossRef CAS PubMed.
- (a) J. A. Palermo, P. B. Flower and A. M. Seldes, Tetrahedron Lett., 1992, 33, 3097 CrossRef CAS; (b) D. Davyt, W. Entz, R. Fernandez, R. Mariezcurrena, A. W. Mombru, J. Saldana, L. Dominguez, J. Coll and E. Manta, J. Nat. Prod., 1998, 61, 1560 CrossRef CAS PubMed.
- S. G. Toske, P. R. Jensen, C. A. Kauffman and W. Fenical, Tetrahedron, 1998, 54, 13459 CrossRef CAS.
- J. Kohno, Y. Koguchi, M. Nisshio, K. Nakao, M. Kuroda, R. Shimizu, T. Ohnuki and S. J. Komatsubara, J. Org. Chem., 2000, 65, 990 CrossRef CAS PubMed.
- Y. Sugie, K. A. Dekker, H. Hirai, T. Ichiba, M. Ishiguro, Y. Shiomi, A. Sugiura, L. Brennan, J. Duignan, L. H. Huang, J. Sutcliffe and Y. Kojima, J. Antibiot., 2001, 54, 1060 CrossRef CAS PubMed.
- (a) R. Tschesche and H. Last, Tetrahedron Lett., 1968, 9, 2993 CrossRef; (b) R. Tschesche, H. Wilhelm and H.-W. Fehlhaber, Tetrahedron Lett., 1972, 13, 2609 CrossRef; (c) Y. N. Han, G.-Y. Kim, H. K. Han and B. H. Han, Arch. Pharmacal Res., 1993, 16, 289 CrossRef CAS.
- R. Shen, C. T. Lin and J. A. Porco, J. Am. Chem. Soc., 2002, 124, 5650 CrossRef CAS PubMed.
- (a) L. Yet, Chem. Rev., 2003, 103, 4283 CrossRef CAS PubMed; (b) K. L. Erickson, J. A. Beutler, J. H. Cardellina II and M. R. Boyd, J. Org. Chem., 1997, 62, 8188 CrossRef CAS PubMed; (c) D. L. Galinis, T. C. McKee, L. K. Pannell, J. H. Cardellina II and M. R. Boyd, J. Org. Chem., 1997, 62, 8968 CrossRef CAS; (d) J. W. Kim, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, J. Org. Chem., 1999, 64, 153 CrossRef CAS PubMed; (e) K. L. Erickson, J. A. Beutler, J. H. Cardellina II and M. R. Boyd, J. Org. Chem., 2001, 66, 1532 CrossRef CAS; (f) K. A. Dekker, R. J. Aiello, H. Hirai, T. Inagaki, T. Sakakibara, Y. Suzuki, J. F. Thompson, Y. Yamaguchi and N. Kojima, J. Antibiot., 1998, 51, 14 CrossRef CAS PubMed; (g) B. Kunze, R. Jansen, F. Sasse, G. Hofle and H. Reichenbach, J. Antibiot., 1998, 51, 1075 CrossRef CAS PubMed.
- C. Bolm and O. Garcia Machenco, Beilstein J. Org. Chem., 2007, 3, 25 CrossRef PubMed.
- (a) S. L. Buchwald, X. Huang and A. Klapars, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef; (b) L. Jiang, G. E. Job, A. Klapars and S. L. Buchwald, Org. Lett., 2003, 5, 3667 CrossRef CAS PubMed.
- ESI†.
- D. R. Williams, M. W. Fultz, T. E. Christos and J. S. Carte, Tetrahedron Lett., 2010, 51, 121 CrossRef CAS PubMed.
- C. Kuang, H. Senboku and M. Tokuda, Tetrahedron, 2005, 61, 637 CrossRef CAS.
- R.-S. Xu, L. Yue and Y.-J. Pan, Tetrahedron, 2012, 68, 5046 CrossRef CAS.
- C. Kuang, Q. Yang, H. Senbokuc and M. Tokudac, Tetrahedron, 2005, 61, 4043 CrossRef CAS.
- F. Zhan and G. Liang, Angew. Chem., Int. Ed., 2013, 125, 1304 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1049804. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra10939f |
| This journal is © The Royal Society of Chemistry 2015 |