
Novel tough and thermally stable cyanate ester resins with high flame retardancy, low dielectric loss and constant based on a phenolphthalein type polyarylether sulfone†
Abstract
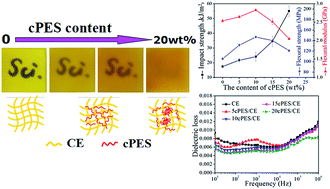
It still remains a big challenge to improve the toughness and flame retardancy of cyanate ester (CE) resin through a simple and effective method without sacrificing its excellent dielectric properties and thermal stability. New modified CE resins were facilely developed through melt-blending with a phenolphthalein type polyarylether sulfone (cPES), and the integrated performances including the reactivity, mechanical, dielectric, thermal and flame retarding properties were systematically investigated. Results show that compared with CE resin, cPES/CE resins have 1.2–3.2 times higher impact strengths according to the concentration of cPES on the basis of maintaining good thermal stability, high modulus, low dielectric constant and loss. Through analyzing thermogravimetric kinetics, cone calorimeter tests, and the structures of chars and pyrolysis gases, it is proven that the addition of cPES into CE resin greatly increases the difficulty of catching fire, reflected by the time to ignition (TTI), fire performance index (FPI) and fire growth index (FGI); while once ignited, the cPES/CE resin exhibits a bigger heat release rate and total heat release. These attractive results demonstrate that cPES is a multi-functional modifier of CE resin. The reason for this was systematically revealed through discussing the influence of cPES on the structure of the crosslinked network.
 Please wait while we load your content...
Please wait while we load your content...

