
Blocking the heat shock response and depleting HSF-1 levels through heat shock protein 90 (hsp90) inhibition: a significant advance on current hsp90 chemotherapies†
Abstract
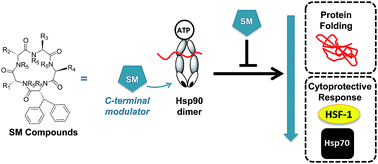
Clinical inhibitors of heat shock protein 90 (hsp90) modulate the N-terminus of the protein, which elicits a cell rescue cascade known as the heat shock response. This cytoprotective mechanism counteracts the impact of hsp90 chemotherapeutic agents. Inhibiting hsp90's activity via the C-terminus does not produce a heat shock response. Herein we report an extensive structure–activity relationship on 41 molecules that are based on the SM class of cyclic pentapeptides. This class of compounds control hsp90's C-terminus function, which induces rapid cell death without activating the heat shock response. We show that modifying single and dual side-chains was one route for producing active molecules. Moving the N-methyl residue around the ring also impact the biological activity of the molecule. Two of the most potent analogues were evaluated for hsp90 inhibitory activity and for their ability to reduce the heat shock response while simultaneously killing cancer cells. In addition, analysis of the most effective molecules in pharmacokinetic studies are described highlighting the compound's potential as a therapeutic drug.
 Please wait while we load your content...
Please wait while we load your content...

