
Structure and thermo-mechanical properties of CTBN-grafted-GO modified epoxy/DDS composites
Raneesh Konnolaa,
Jinu Jojib,
Jyotishkumar Parameswaranpillaic and
Kuruvilla Joseph*a
aDepartment of Chemistry, Indian Institute of Space Science and Technology, Thiruvananthapuram-695547, Kerala, India. E-mail: kuruvilla@iist.ac.in; kjoseph.iist@gmail.com; Tel: +91 471 2568541
bDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal-462066, Madhya Pradesh, India
cDepartment of Polymer Science and Rubber Technology, Cochin University of Science and Technology, Cochin-682022, Kerala, India
First published on 10th July 2015
Abstract
Carboxyl terminated poly(acrylonitrile-co-butadiene) (CTBN) is grafted on to graphite oxide (GO) to prepare GCTBN in order to improve the dispersion and interfacial bonding between GO and epoxy resin in an epoxy/DDS system. GCTBN was characterized by FTIR, XPS, Raman spectroscopy, XRD, TEM, TOM (morphology) and TGA. All these studies reveal the grafting of CTBN with GO. The thermal stability of GCTBN was found to improve considerably. The TEM micrograph of epoxy/GCTBN reveals an excellent dispersion of GCTBN in the epoxy matrix. Tensile strength (ca. 25%), tensile modulus (ca. 34%), tensile elongation (ca. 10%), and fracture toughness (ca. 128%) improved remarkably for GCTBN modified epoxy matrix. SEM micrographs reveal no sheet pull out for GCTBN modified epoxy, due to the complete wetting of GCTBN by the epoxy matrix. This confirms effective sheet/matrix interfacial bonding for the GCTBN modified epoxy matrix. Moreover, the viscoelastic properties reveal a very high modulus and improved Tg for the epoxy/GCTBN when compared with the neat crosslinked epoxy.
1. Introduction
Epoxy resin is an important thermosetting matrix with good stiffness, thermal resistance, chemical resistance and long pot life period. On the other hand, cured epoxies are highly brittle, which limits their utility in many composite applications. The toughness can be drastically improved by the addition of a second phase. From the literature, it can be seen that reactive rubbers, thermoplastics and block copolymers can be used as an excellent tougheners for epoxy systems.1–6 Several recent studies point towards the use of nano fillers such as CNTs, graphene, CNF, clay, SiO2, TiO2 etc. as modifiers for epoxy resins.7–13The major problem associated with polymer nanocomposites is the poor dispersion of the filler in the polymer matrix. Generally nanoparticles have a tendency for agglomeration because of the weak van der Waals force of attraction. In fact, GO has a strong tendency of aggregation inside epoxy matrix which limits the equal distribution of load into matrix. Chemical modifications of GO sheets are an effective way to improve interfacial interaction between the GO sheets and the epoxy matrix, which in turn leads to better filler dispersion, and enhanced mechanical performance in the nanocomposites.14–17 Naebe et al. functionalized thermally reduced graphene nanoplatelets via Bingel reaction18 to evaluate the effect of functionalization on the dispersion status and interface in the graphene/epoxy composites. A good improvement in fracture toughness was observed in the work. In an another study, Park et al.19 investigated the toughening behavior of epoxy nanocomposites using amine terminated poly(acrylonitrile-co-butadiene) functionalized GO as reinforcing filler and they found a significant improvement in toughness at very small filler loadings. Similarly, Guan et al.20 introduced amine groups of polyetheramine (PEA) with different molecular lengths onto the GO surface, and studied sheet/matrix interfacial interaction between filler and epoxy matrix to understand the influence of different interphase structures on the mechanical properties of resulting nanocomposites. Wang et al.21 synthesized polyphosphamide (PPA) and covalently grafted it onto the surface of graphene nanosheets (GNSs). These modified sheets were incorporated into epoxy resins (EPs) to obtain a novel flame retardant nanocomposite. Tang and coworkers were able to achieve an electrical conductivity of nearly 11 orders of magnitude higher than that of neat epoxy by the addition of 2.7 vol% of polyetheramine functionalized GOs.22 The above studies reveal the potential behind the chemical modification of GO sheets for the improvements in properties at low filler loading, provides opportunity to produce cost effective high performance epoxy graphene composites.
The present work is focused on the chemical modification of GOs with a conventional liquid rubber CTBN, in an attempt to achieve high dispersion and enhanced interaction in an epoxy matrix and hence to prepare composites with improved thermo-mechanical properties. Grafting of GO sheets with CTBN can create a soft interface between filler and matrix which can result in a better load transfer from matrix to filler. To date, no systematic study of grafting CTBN rubber onto GO to improve the compatibility and performance in the epoxy composites is available in the literature. In this work, CTBN grafted GO (GCTBN) is prepared, and used as a modifier for epoxy resin. We investigated thermal, viscoelastic and mechanical performance of GCTBN modified epoxy nanocomposites and thereby evaluated the effect of chemical modification on the dispersion and interfacial interaction in the resulting composites.
2. Materials and methods
2.1 Materials
Graphite was supplied by Anthracite industries, USA, concentrated sulphuric acid (H2SO4, 98%), concentrated hydrochloric acid (HCl, 35%), potassium permanganate (KMnO4), hydrogen peroxide (H2O2, 30%) and acetone were purchased from Merck India Pvt Ltd, Bangalore India. The polymer matrix used in the present study was epoxy resin Lapox ARL-135 based on diglycidyl ether of bisphenol A (DGEBA) (epoxy equivalent 187 g per eq.) and diaminodiphenyl sulfone (DDS) hardener under the commercial name Lapox K10 and was purchased from Atul India private limited, Gujarat. Dimethyl formamide (DMF) purchased from Spectrochem, India. Triphenylphosphine (TPP, 99% MW-262.92 g mol−1) and poly(acrylonitrile-co-butadiene) dicarboxyl terminated (CTBN, MW-3600 g mol−1) purchased from Sigma Aldrich, Bangalore, India. All the chemicals were used as received without further purification.2.2 Modifications on graphene oxide
The graphite was first exfoliated to form GO using the improved method reported by Tour group.23 In a typical process a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) was added to a mixture of expandable graphite (3.0 g, 1 wt equiv.) and KMnO4 (18.0 g, 6 wt equiv.). The reactants were heated to 50 °C and stirred for 12 h. The mixture was cooled to room temperature and it was kept in an ice bath. To this solution, 400 mL deionised water was added dropwise with stirring for 30 minutes. 30% H2O2 was slowly added into the mixture until the solution turned bright yellow. The resulting yellowish brown mixture was centrifuged and the solid material was then washed in succession with 200 mL of water, 200 mL of 30% HCl and 200 mL of ethanol. After this multiple wash, it was coagulated with 200 mL of ether. The solid GOs obtained after the evaporation of ether, was vacuum-dried overnight at room temperature. For the preparation of GCTBN, 500 mg of GOs was sonicated in DMF for 30 minutes. In a separate beaker, 2 g CTBN in 20 mL DMF solution was sonicated for 30 minutes and this solution was added to GO solution with mechanical stirring. 0.50 wt% TPP was added and the solution was heated at 125 °C for 36 h. The suspension was filtered and washed with DMF followed by acetone. The black powder obtained was dried well and powdered. A schematic illustration of the reaction is given in Fig. 1(a).
:1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) was added to a mixture of expandable graphite (3.0 g, 1 wt equiv.) and KMnO4 (18.0 g, 6 wt equiv.). The reactants were heated to 50 °C and stirred for 12 h. The mixture was cooled to room temperature and it was kept in an ice bath. To this solution, 400 mL deionised water was added dropwise with stirring for 30 minutes. 30% H2O2 was slowly added into the mixture until the solution turned bright yellow. The resulting yellowish brown mixture was centrifuged and the solid material was then washed in succession with 200 mL of water, 200 mL of 30% HCl and 200 mL of ethanol. After this multiple wash, it was coagulated with 200 mL of ether. The solid GOs obtained after the evaporation of ether, was vacuum-dried overnight at room temperature. For the preparation of GCTBN, 500 mg of GOs was sonicated in DMF for 30 minutes. In a separate beaker, 2 g CTBN in 20 mL DMF solution was sonicated for 30 minutes and this solution was added to GO solution with mechanical stirring. 0.50 wt% TPP was added and the solution was heated at 125 °C for 36 h. The suspension was filtered and washed with DMF followed by acetone. The black powder obtained was dried well and powdered. A schematic illustration of the reaction is given in Fig. 1(a).
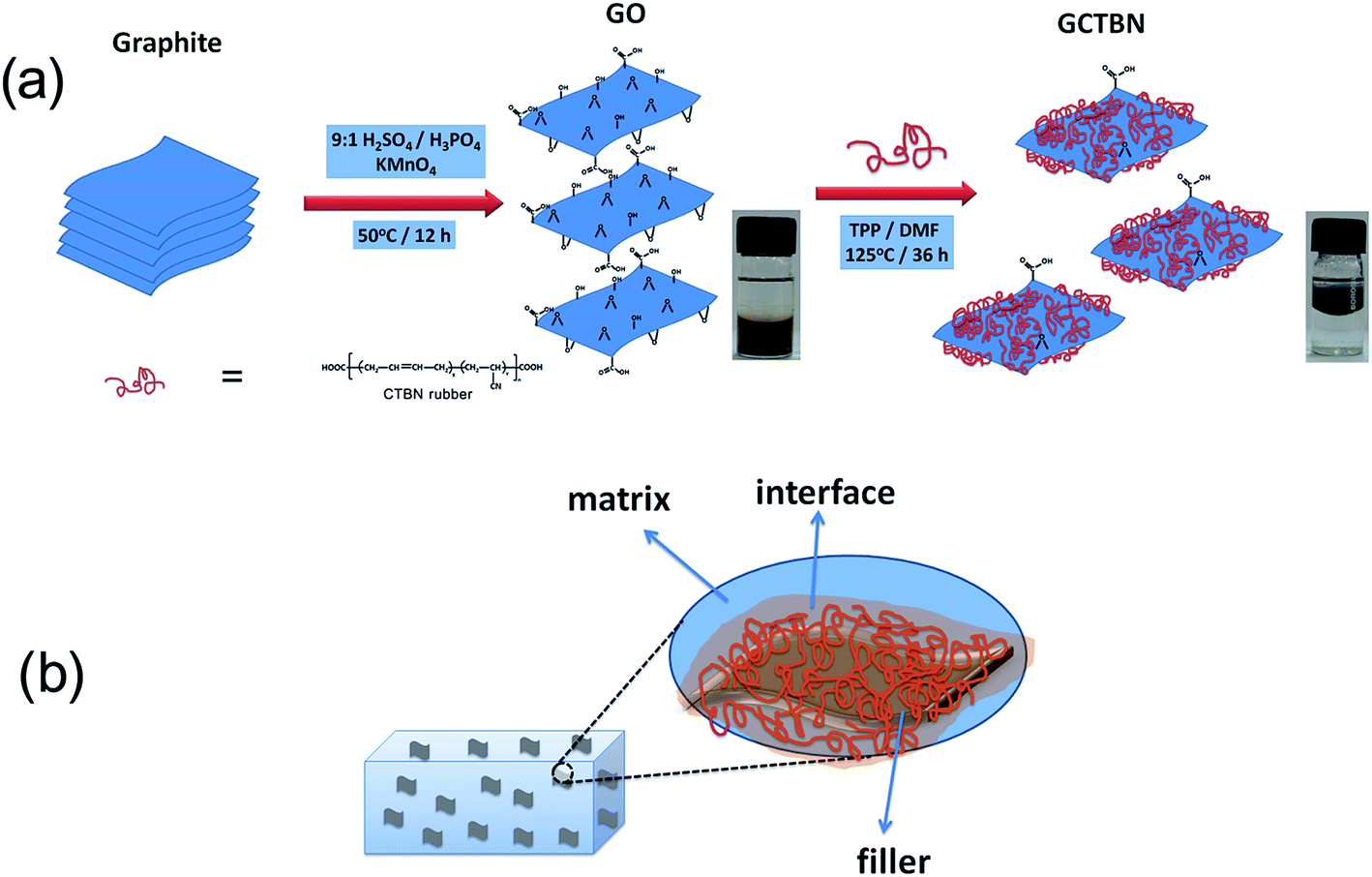 | ||
| Fig. 1 Schematic showing (a) the preparation of GO and GCTBN and (b) the interface between the epoxy matrix and GCTBN filler. | ||
2.3 Preparation of GO–epoxy nanocomposite
Epoxy composites with different GO loadings were prepared by the following procedure. Required amounts of GOs (0.2, 0.4, 0.6 and 0.8 wt% with respect to DGEBA) was initially dissolved in a mixture of acetone by sonication for 15 minutes and mixed the solution with DGEBA. The solution was again sonicated for 15 minutes to obtain a black suspension. Acetone was evaporated off by heating at 50 °C for 1 h. The trace amount of solvent was removed by keeping in a vacuum oven. It was then added to molten hardener (35 g/100 g DGEBA), stirred for 10 minutes and degassed for 10 minutes until there was no trace of trapped bubbles. The mixture was poured into a preheated mould and cured for 4 h at 180 °C. Post curing was done at 200 °C for an hour. The same procedure was followed for the preparation of modified GO/epoxy composite. The preparation process of epoxy composites filled with GOs is shown in Fig. 2.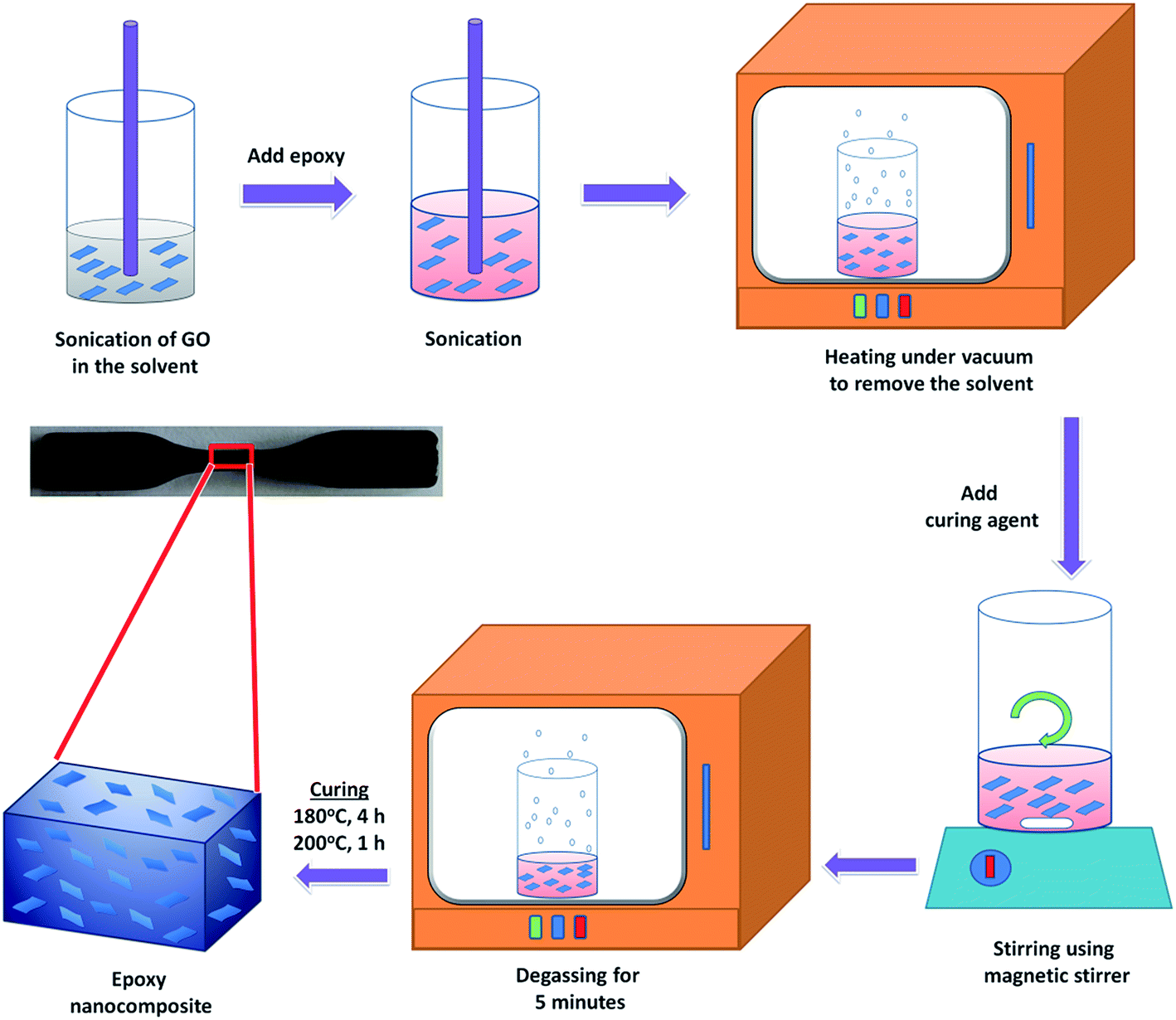 | ||
| Fig. 2 Schematic illustration of the preparation process of epoxy nanocomposite. | ||
2.4 Characterization techniques
Fourier transform infrared spectroscopy (FT-IR) analysis was carried out using a Perkin Elmer System series 100 spectrophotometer in a frequency range of 4000–500 cm−1 with a spectral resolution of 4 cm−1 to identify the functionalization of GO. XPS was carried out with a Kratos Axis Ultra DLD spectrometer, using Al Kα excitation radiation. The high-resolution transmission electron microscopy (HRTEM) was conducted using JEOL JEM-2100 with an acceleration voltage of 200 kV being equipped with an EDX spectrometer. Sliced thin sections of GO/epoxy composites with a thickness of about 60–80 nm, prepared by ultra-microtomy, were used to take the TEM images of the composites. The crystal phase of the samples was measured using X-ray diffractometry (PANalytical 3 kW X'pert PRO X-ray diffractometer) using Cu Kα (k = 1.5406 Å) radiation source operating at a voltage of 45 kV and 300 mA of electric current. The scanning was taken from 5 to 80° (2 h). Raman spectra were recorded from 100 to 3000 cm−1 on a Raman spectrometer (INVIA, England) with a 514 nm argon ion laser. Thermal stability of nanocomposites was analyzed using a Thermogravimetric analyzer (Q-50, TA Instruments, USA). The samples were heated from ambient to 800 °C at a ramp rate of 10 °C min−1. Rheological analysis was done using Modular Rheometer (MCR102, Anton Paar, USA), using a 50 mm parallel plate assembly at room temperature. Optical microscopy analysis was carried out with Leica DM1000 LED (Leica Microsystems, Germany) in transmitted light configuration. The analysis was done on a small droplet of the epoxy suspension placed on a microscope glass. Dynamic Mechanic Analysis (DMA) was performed on a DMA 8000, operating in the single cantilever mode at an oscillation frequency of 1 Hz. Data was collected from room temperature to 250 °C at a scanning rate of 2 °C min−1. The sample specimens were cut into rectangular bars measuring 50 mm × 5 mm × 2 mm. Tensile tests were performed with dumbbell shaped specimens using an Instron model 5900 tensile tester at a crosshead speed of 1 mm min−1 as per ASTM standard D638. The results are the average of at least five measurements. Fracture toughness of the sample was measured using UTM (Instron 5900, Instron, USA) at a crosshead speed of 10 mm min−1 (as per ASTM standard D5045). Single edge notch specimens of 46 mm × 6 mm × 3 mm (span length = 24 mm) were used to measure the fracture toughness of the epoxy nanocomposites. A notch of 2.7 mm was made at one edge of the specimen. A natural crack was made by pressing a fresh razor blade into the notch. The fracture toughness was expressed as stress intensity factor (KIC) calculated using equation
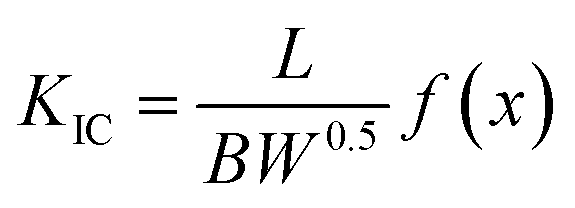 | (1) |
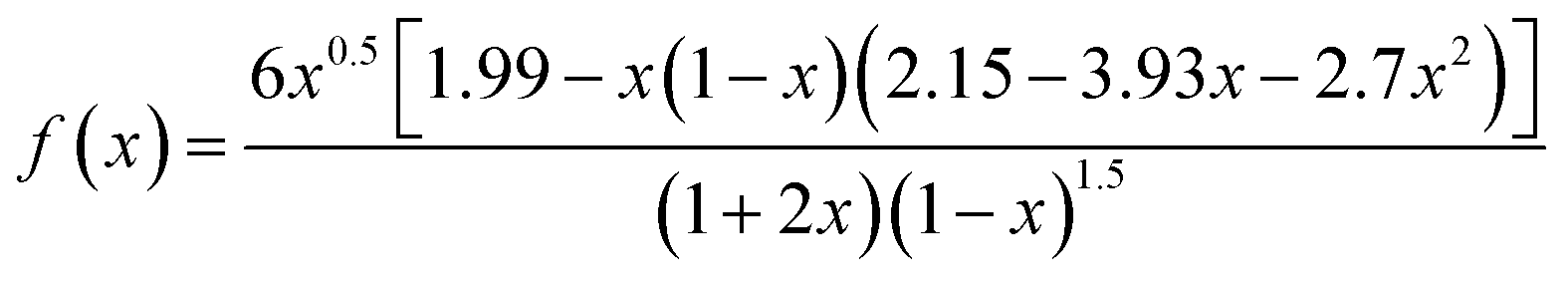 | (2) |
The fracture surfaces of the samples were investigated using field emission SEM (FE SEM, FEI Quanta FEG200) at an accelerating voltage of 20 kV, and the fracture surfaces were coated with a conductive layer of gold.
3. Results and discussions
3.1 Characterization of grafting of CTBN on GO
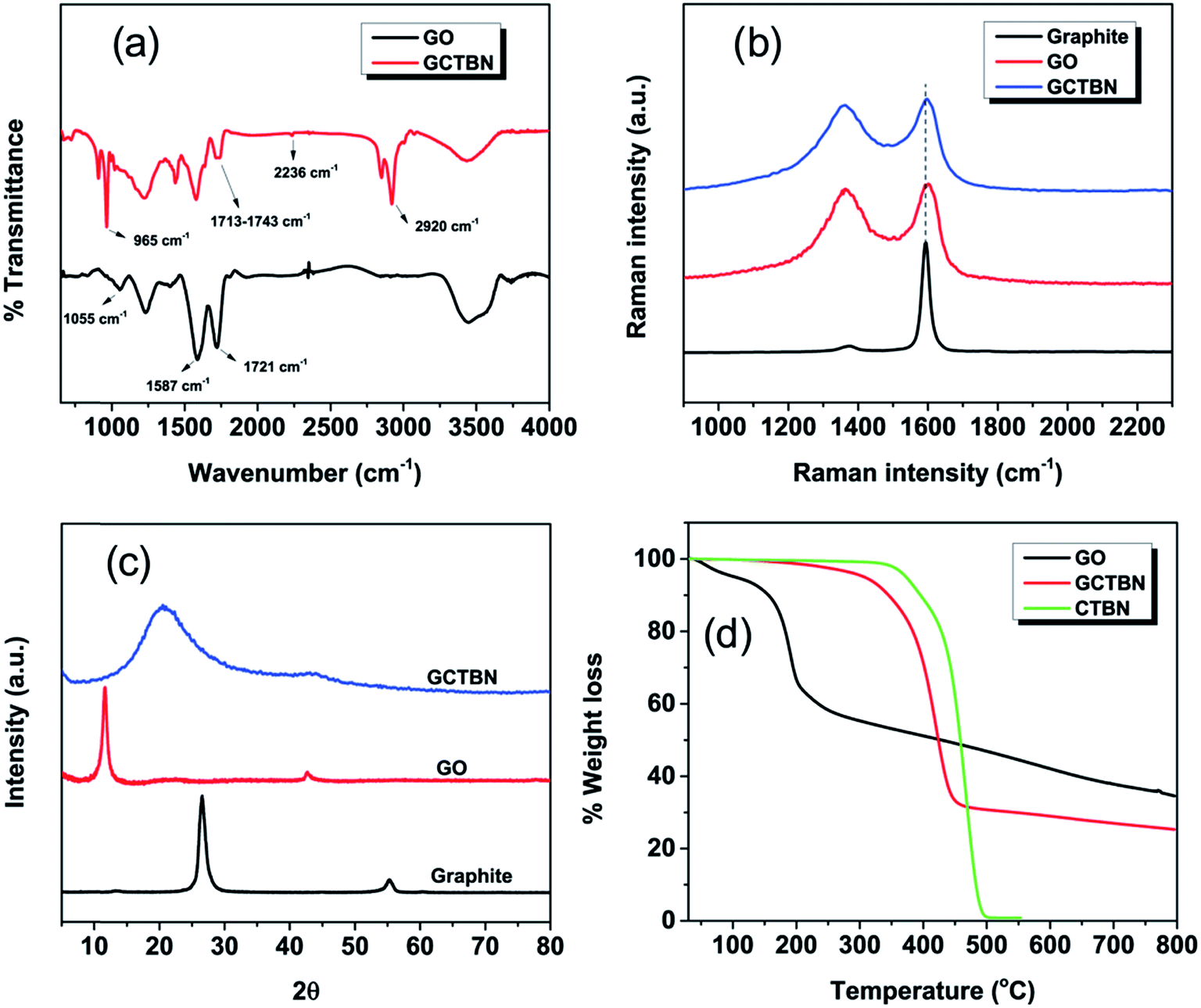
The FTIR spectra of GO and GCTBN were recorded to obtain information about the structural changes originated during the grafting process (Fig. 3(a)). The characteristic absorption bands of GOs in the FTIR spectrum were observed at 1721 cm−1, 1055 cm−1, 1587 cm−1 and 3446 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations from carbonyl and carboxylic groups, C–O–C stretching from epoxy groups, CC in aromatic ring and –O–H stretching frequency of hydroxyl groups respectively.20 After the grafting with CTBN, the FTIR spectrum of GO is significantly changed, with the appearance of new and more intense peaks. The new characteristic peaks at 965 cm−1, 2236 cm−1 and 2920 cm−1 appearing in the FTIR spectrum of GCTBN, corresponds to the C–H out of plane bending vibration of 1,4 trans olefin in CTBN, stretching vibration of C
O stretching vibrations from carbonyl and carboxylic groups, C–O–C stretching from epoxy groups, CC in aromatic ring and –O–H stretching frequency of hydroxyl groups respectively.20 After the grafting with CTBN, the FTIR spectrum of GO is significantly changed, with the appearance of new and more intense peaks. The new characteristic peaks at 965 cm−1, 2236 cm−1 and 2920 cm−1 appearing in the FTIR spectrum of GCTBN, corresponds to the C–H out of plane bending vibration of 1,4 trans olefin in CTBN, stretching vibration of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and the stretching vibration of C–H, respectively.24 Furthermore, the intensity of peak at 1055 cm−1 corresponding to C–O–C of epoxy groups is reduced drastically in the spectrum of GCTBN and the peak corresponding to CO stretching vibration is broadened (1713–1743 cm−1), indicating the formation of OC–O ester bond due to the chemical reaction of CTBN to GO surface via nucleophilic substitution reaction between the carboxyl groups of CTBN and the epoxy groups of GO. All these results confirm the successful modification of CTBN with GO.
N and the stretching vibration of C–H, respectively.24 Furthermore, the intensity of peak at 1055 cm−1 corresponding to C–O–C of epoxy groups is reduced drastically in the spectrum of GCTBN and the peak corresponding to CO stretching vibration is broadened (1713–1743 cm−1), indicating the formation of OC–O ester bond due to the chemical reaction of CTBN to GO surface via nucleophilic substitution reaction between the carboxyl groups of CTBN and the epoxy groups of GO. All these results confirm the successful modification of CTBN with GO.
 | ||
| Fig. 3 (a) FTIR, (b) Raman spectra (c) XRD and (d) TGA of GO and GCTBN. | ||
The Raman spectra of graphite, graphite oxide and GCTBN are shown in Fig. 3(b). Graphite is usually characterized by two main features, the G band at 1593 cm−1 resulting from first order scattering of the in plane vibration of E2g photon of sp2 carbon atoms of graphitic lattice and the D band at 1375 cm−1 arising from a breathing mode of κ-point photons of A1g symmetry.22 Quantification of the intensity ratio of the D band to G band (i.e., ID/IG) reveals the extent of defects created by the chemical treatment. The ID/IG ratio of graphite is very small (0.05). After the oxidation, the two bands broaden and shift to high frequency accompanied by an increased ID/IG value (2.12), indicating the distortion of the bonds and destruction of symmetry due to the reduction in size of the in plane sp2 domains caused by the extensive oxidation. However, the G peak of the GCTBN shifts from 1601 to near 1596 cm−1, getting close to that of natural graphite (1593 cm−1) implying restoration of the graphitic sp2 network.25 Compared to the as-produced GO, GCTBN sheets show slight increase in the ID/IG values (from 2.12 to 2.34), which further confirms the formation of covalent bonds between the GO and the CTBN molecules.
Fig. 3(c) represents the XRD pattern of graphite, GO and GCTBN. Graphite show a characteristic diffraction peak at 26.5° representing the (002) reflection peak corresponding to an interlayer distance of 0.34 nm.26 After the oxidation of graphite, GO shows a diffraction peak at lower diffraction angle at 11.63° degree indicating the increase of interlayer spacing due to presence of oxygen functional group at the surfaces and edges as a result of vigorous oxidation process. Or in other words (002) reflection peak disappeared indicating that the graphene sheets are disordered. XRD pattern of GCTBN shows a weak and broad peak from 11–30 centered at 20.5° indicating the disappearance of the long-term ordering graphitic structure and the crystalline organization of sheets were affected by the presence of rubber.
Thermal gravimetric curves of graphite, GO and GCTBN are shown in Fig. 3(d). From the figure, it is understood that GO is highly unstable and has an initial mass loss around 5%, below 100 °C due to the evaporation of absorbed water,27,28 and the major weight loss of 40% around 100–300 °C is ascribed to the pyrolysis of the labile oxygen-containing functional groups, yielding CO, CO2 and steam.29 The percent weight of GO further decreased up to 800 °C due to the degradation of carbon backbone. The functionalization and reduction of GO improves the thermal stability of the reinforcing filler. This is evident from the fact that a decomposition of only 4.4% is observed in the temperature region of 100–300 °C. TGA curve of GCTBN shows a major decomposition of 67% in the temperature range of 300–500 °C which could be attributed to the decomposition of CTBN polymer chains that were grafted on the GO sheets. The color change from brown to black and the reduction in intensities of oxygen functional groups in FTIR, XPS and TGA curves indicates the partial reduction of GO after the polymer grafting. This phenomenon of reduction of GOs during functionalization is observed elsewhere.22,30–33
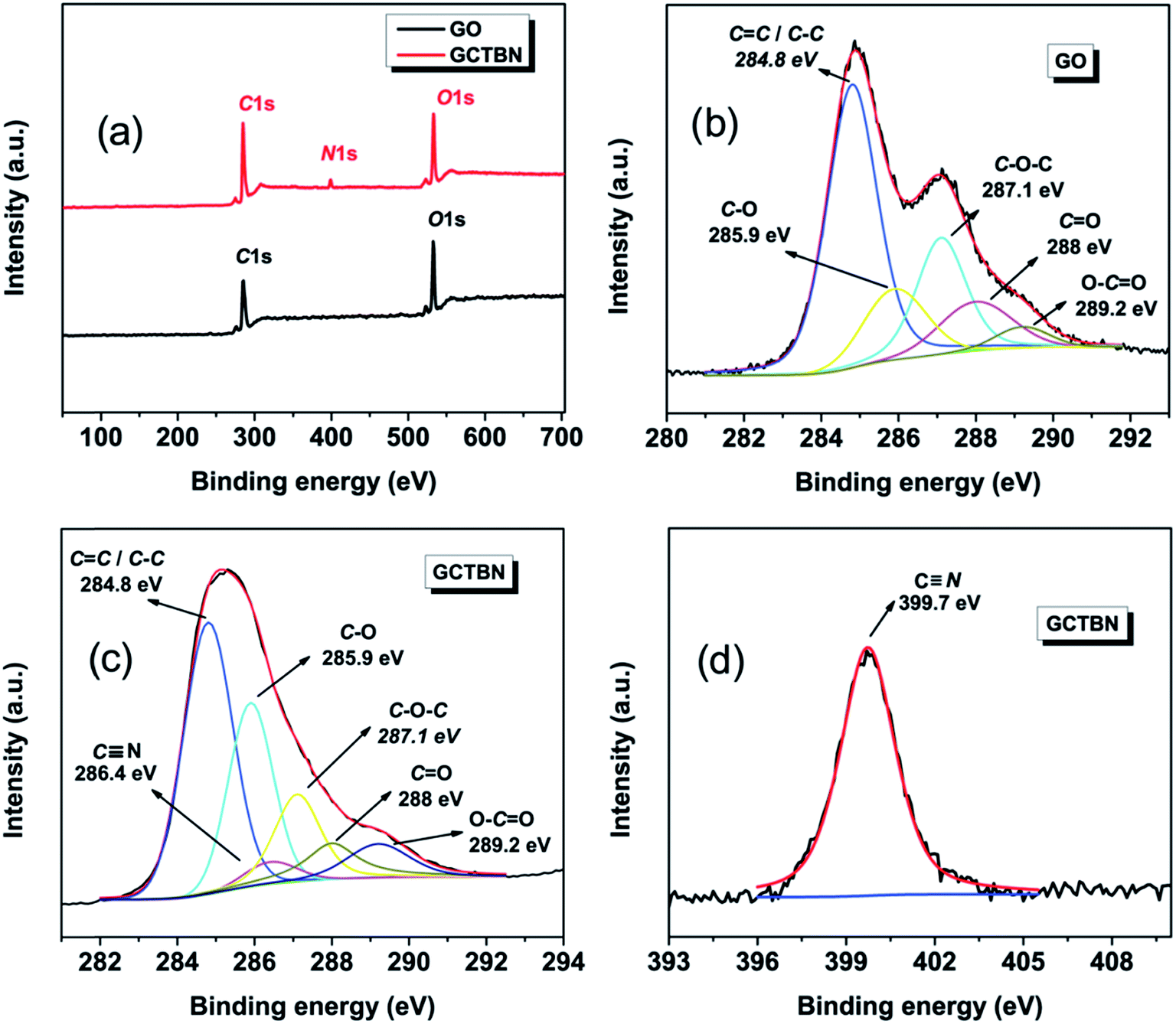
Analysis of the XPS spectra provides clear evidence of the fact that the GOs were chemically modified. The XPS survey spectra of (a) GO and GCTBN, and higher resolution C1s spectra of (b) GO, (c) GCTBN and (d) N1s spectra of GCTBN are shown in Fig. 4. Compared with GO, the survey of GCTBN shows the presence of N1s originating from nitrile group of CTBN, indicating the chemical grafting of CTBN chains onto the surface of the GO sheets. The C1s core level spectra of GOs shows peaks at 284.8 eV (C–C/CC), 285.9 eV (C–OH), 287.1 eV (C–O–C/epoxide group), 288.0 eV (CO), and 289.2 eV (O–CO), respectively.34,35 Although the C1s XPS spectrum of the GCTBN (Fig. 4(c)) also exhibits the same oxygen functionalities, their peak intensities are much smaller than those in GO indicating partial reduction of GO during the reaction with CTBN. In addition, the area of the peak at 287.1 eV is decreased drastically in the C1s spectrum of GCTBN (Fig. 4(c)) indicating that the reaction has happened between the epoxide group of GO and carboxyl groups present in CTBN. An additional peak at 286.4 eV in the C1s high resolution and peak at 399.7 eV in the N1s spectra is arising from the CN group in CTBN.36 These above mentioned XPS results further demonstrates that GO is successfully functionalized by CTBN molecules, which is in agreement with FTIR results.
 | ||
| Fig. 4 (a) XPS survey spectra of GO and GCTBN, and high resolution C1s spectra of (b) GO, (c) GCTBN and (d) N1s spectra of GCTBN. | ||
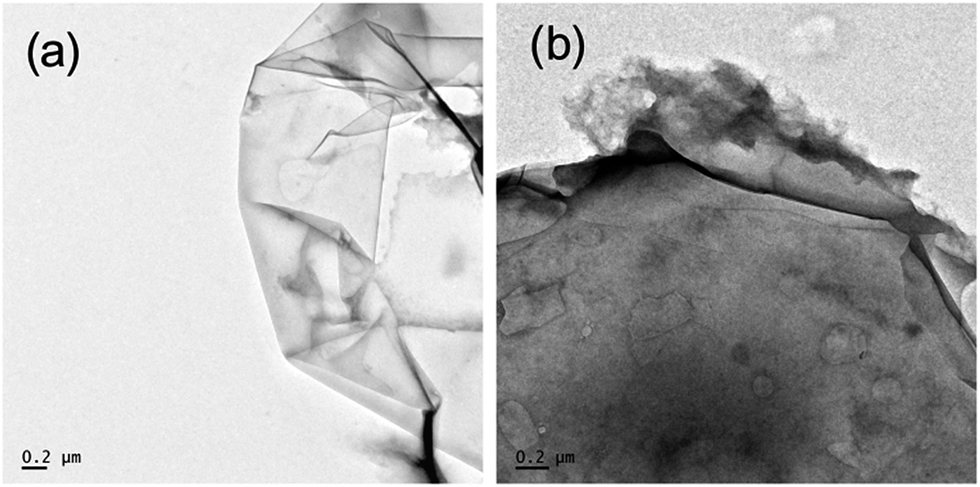
The morphology and structure of GO and GCTBN were determined by TEM analysis. The TEM image of the prepared GO sheets shows a thin sheet-like two-dimensional structure with a diameter of several micrometers. GO sheets contain a lot of wrinkles due to the presence of epoxy and hydroxyl functional groups within the graphene sheets.14 After the polymer functionalization, GCTBN sheets exhibited a rougher and thicker structure and a thin polymer layer seems to be observed surrounding the sheets, as shown in Fig. 5, suggesting the successful grafting of CTBN on GO through nucleophilic attack by carboxyl group of CTBN to the epoxy groups of GO.
 | ||
| Fig. 5 TEM images of (a) GO and (b) GCTBN. | ||
3.2 Dispersion behavior of nanosuspension and composite
 | ||
| Fig. 6 TOM images of epoxy nanosuspensions containing (a) 0.4 wt% GO, (b) 0.8 wt% GO, (c) 0.4 wt% GCTBN and (d) 0.8 wt% GCTBN. Insets show high magnification images. | ||
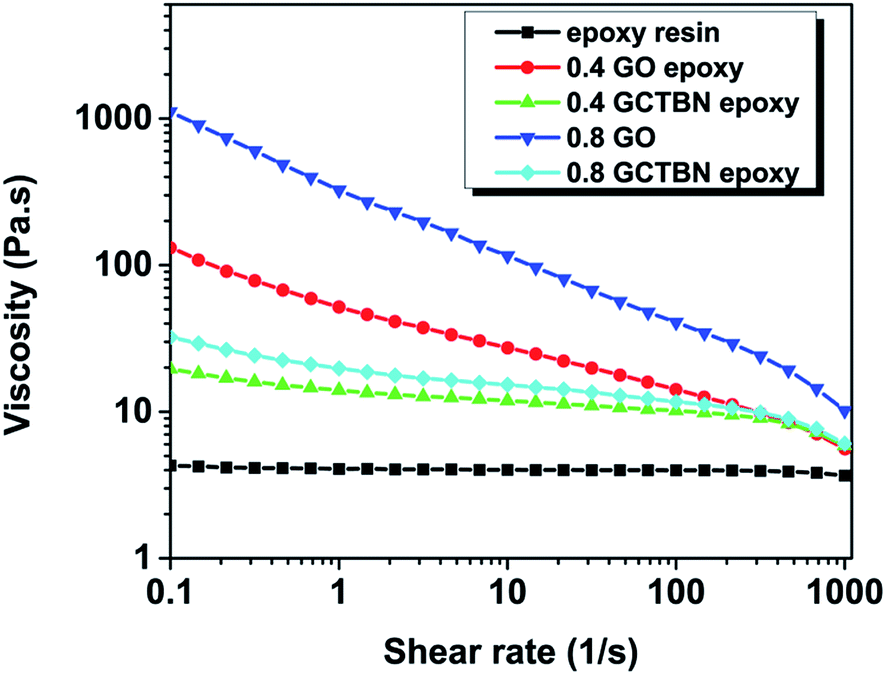 | ||
| Fig. 7 Variation of viscosity with shear rate for epoxy nano suspension of GO and GCTBN. | ||
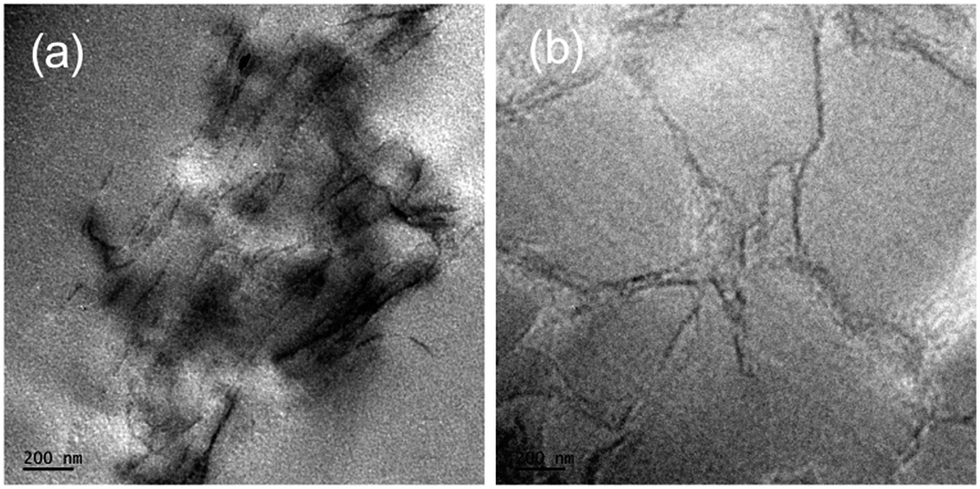 | ||
| Fig. 8 TEM images of epoxy nanocomposites containing (a) 0.6 wt% GO and (b) 0.6 wt% GCTBN. | ||
3.3 Tensile strength
The tensile properties for neat epoxy and its composites with different filler loadings of GOs and GCTBN are summarized in Fig. 9 and corresponding values are given in Table 1. From the table, the addition of GOs improved the tensile properties. The tensile strength increases maximum by ca. 22% by the addition of 0.4 wt% GOs. Similarly, the tensile modulus and tensile elongation shows a maximum increase of ca. 16% and ca. 36% respectively with the addition of 0.6 wt% GOs. The increase in tensile modulus represent the improved stiffness, on the other hand the increase in elongation at break represent the improved ductility. This means that the modification of epoxy with small amount of GOs (0.4 to 0.6 wt% of GOs) is an ideal way to improve the properties of epoxy systems. In fact this improvement in properties make the GO based composites attractive for many industrial applications.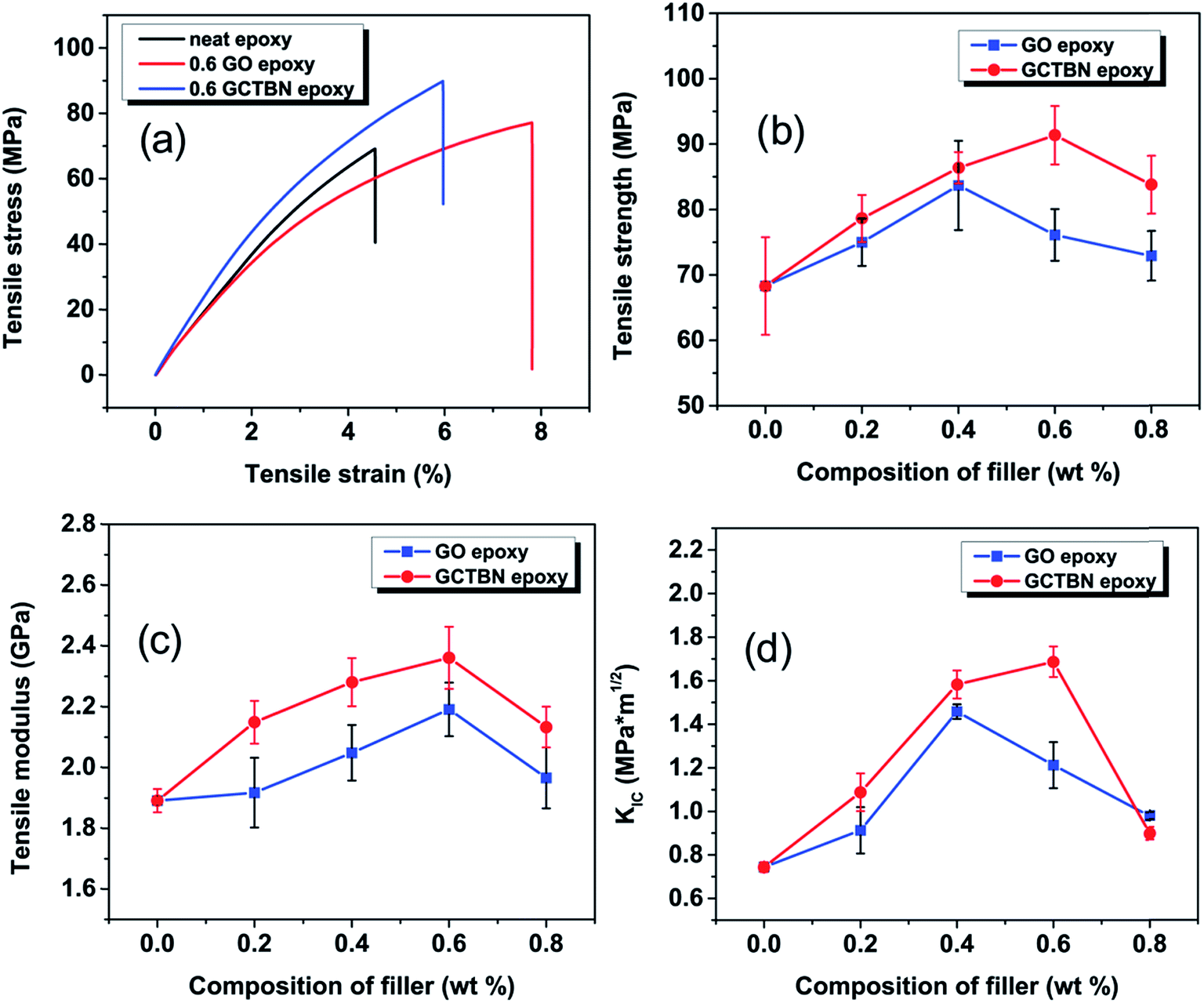 | ||
| Fig. 9 (a) Representative stress–strain curves (b) tensile strength (c) tensile modulus and (d) fracture toughness of epoxy nanocomposites containing 0.6 wt% GO and 0.6 wt% GCTBN. | ||
| SI no. | Sample ID | KIC (MPa m1/2) | Tensile strength (MPa) | Tensile modulus (GPa) | Elongation at break (%) |
|---|---|---|---|---|---|
| 1 | Neat epoxy | 0.74 ± 0.02 | 68.4 ± 1.7 | 1.89 ± 0.04 | 5.66 ± 0.63 |
| 2 | 0.2 wt% GO | 0.91 ± 0.10 | 75 ± 3.6 | 1.92 ± 0.11 | 6.01 ± 0.88 |
| 3 | 0.4 wt% GO | 1.46 ± 0.03 | 83.7 ± 6.8 | 2.05 ± 0.09 | 7.23 ± 1.45 |
| 4 | 0.6 wt% GO | 1.21 ± 0.10 | 76.1 ± 3.9 | 2.19 ± 0.09 | 7.69 ± 0.99 |
| 5 | 0.8 wt% GO | 0.98 ± 0.02 | 72.9 ± 3.8 | 1.97 ± 0.10 | 5.58 ± 0.67 |
| 6 | 0.2 wt% GCTBN | 1.09 ± 0.09 | 78.6 ± 3.6 | 2.15 ± 0.07 | 8.27 ± 0.11 |
| 7 | 0.4 wt% GCTBN | 1.58 ± 0.06 | 86.4 ± 2.4 | 2.28 ± 0.08 | 6.05 ± 0.40 |
| 8 | 0.6 wt% GCTBN | 1.69 ± 0.07 | 91.4 ± 4.5 | 2.36 ± 0.10 | 6.23 ± 1.27 |
| 9 | 0.8 wt% GCTBN | 0.90 ± 0.03 | 83.8 ± 4.4 | 2.13 ± 0.07 | 6.96 ± 0.88 |
The composites containing the GCTBN exhibit better tensile strength, and modulus values than their GO counterparts. Maximum tensile strength and modulus are observed for the composites with 0.6 wt% GCTBN. For epoxy composite with 0.6 wt% GCTBN, the tensile modulus, strength and elongation increased by ca. 25% (2.36 ± 0.16 GPa), and ca. 34% (91.4 ± 4.3 MPa), and ca. 10% (6.23 ± 1.27) respectively with respect to neat epoxy system. From these results, it is clear that the reinforcing capability of GCTBN was better than that of GOs. The mechanical properties of the epoxy nanocomposites depend on the proper dispersion of fillers in the polymer matrix along with a good interaction between the reinforcement and the polymer. After grafting of GO with CTBN, the fine dispersion and exfoliation of GCTBN sheets as well as the strong interfacial interaction between GO and epoxy due to the presence of soft polymer at the interface, favors proper stress transfer between the matrix and reinforcing filler and thus result in a significant enhancement in the tensile properties. This is schematically represented in Fig. 1(b).
3.4 Fracture toughness of epoxy composite
Ability of a material to resist fracture is described as fracture toughness and is expressed in critical stress intensity factor (KIC). KIC values of neat epoxy, GO, and GCTBN epoxy nanocomposites are given in Table 1. Addition of GO and GCTBN into the epoxy matrix resulted in an improvement in fracture toughness (Fig. 9(d)). By the addition of 0.4 wt% GOs, KIC increased from 0.74 ± 0.02 MPa m1/2 to 1.46 ± 0.03 MPa m1/2 an increase of ca. 97% improvement. This is followed by a decrease in the value of KIC at higher GO concentrations due to the presence of agglomerates and defects. GCTBN modified epoxy composites shows higher fracture toughness values than GO epoxy nanocomposites with highest fracture toughness of 1.69 ± 0.07 MPa m1/2 with an improvement of 128% compared to neat epoxy. The decrease in value of KIC at 0.8 wt% in the case of GCTBN modified epoxy might be due to the presence of agglomerates.To evaluate the dispersion and interfacial behavior of the composites, the fracture surface obtained after the fracture toughness test was evaluated using HRSEM (Fig. 10). Fracture surface of neat epoxy shows smooth mirror like pattern which suggests poor absorption of energy during crack propagation which results in brittle fracture. Considerable difference is noticed between the failure surface of neat epoxy and that of GO epoxy composite system. The fracture surfaces of the composites became very rough with the inclusion of GOs. These changes are attributed to the crack deflection and pinning created by the addition of the rigid GO sheets. The total fracture surface area of the system is increased as a result of incorporation of GOs resulting in greater energy absorption as compared to that of the unfilled polymer. The SEM image of the epoxy composites containing higher loading of GOs shows non-uniform dispersion due to aggregated GOs. Aggregates of GO with size of several microns were observed at the surface of 0.8 wt% GO modified epoxy composite (shown as red circle in Fig. 10(c)). This indicates the breakdown of filler/matrix interface or in other words represents poor interactions between matrix and particle.13 Such aggregates of GO sheets and poor filler/matrix interface would cause stress concentrations during the fracture process. These stress concentrations may facilitate failure during the fracture test. The fracture surface of GCTBN modified epoxy composite reveals a different surface morphology, and is relatively coarser than the composites containing GO. The rougher surfaces signify much more energy absorption. Moreover, SEM micrographs reveal no sheet pull out, which means that graphene surface is fully wet by the epoxy matrix. This indicates that the sheet/matrix interfacial bonding is effectively improved after doing CTBN surface functionalization.18 Since GCTBN produced a stronger interface with the matrix than GO, GCTBN was able to carry a higher level of loading upon fracture, as indicated by the excellent fracture features. As in the case of GO modified epoxy composites, a higher loading of GCTBN in the system resulted in the agglomeration of sheets resulting in a decrease in the KIC values.
 | ||
| Fig. 10 FESEM images of fractured surface of sample: (a) neat epoxy (b) 0.6 wt% GO (c) 0.8 wt% GO (d) 0.6 wt% GCTBN and (e) 0.8 wt% GCTBN. | ||
3.5 Dynamic mechanical analysis
DMA was carried out to understand the viscoelastic behavior over a wide range of temperatures for neat epoxy, GO modified epoxy and GCTBN modified epoxy. Fig. 11(a) shows a comparison of the storage modulus (E′) of neat epoxy and epoxy with 0.6 wt% of GOs and GCTBN in the temperature range 30–250 °C. The storage modulus of neat epoxy system was improved significantly with the incorporation of GO sheets. The storage modulus at the glassy region and rubber region are given in Table 2. From the table the storage modulus of neat epoxy at 30 °C is 1995 MPa, while that of GO and GCTBN modified epoxy composites are 2319 and 2476 MPa, respectively. These values were ca. 15% and ca. 25% higher than those obtained for the neat epoxy system. The increased stiffness is due to the high modulus of graphene platelets dispersed in the epoxy matrix. On the other hand, the storage modulus is maximum for GCTBN modified epoxy composites. This is due to the strong interfacial interaction between the filler and matrix after chemical modification with rubber, which reduces the mobility of the local matrix around the sheets. All the composites show two inflection points, one at 60 °C, due to lower cross-link density sites in the epoxy network and the other at the glass transition temperature (Tg) of the cross-linked epoxy system at around 200 °C.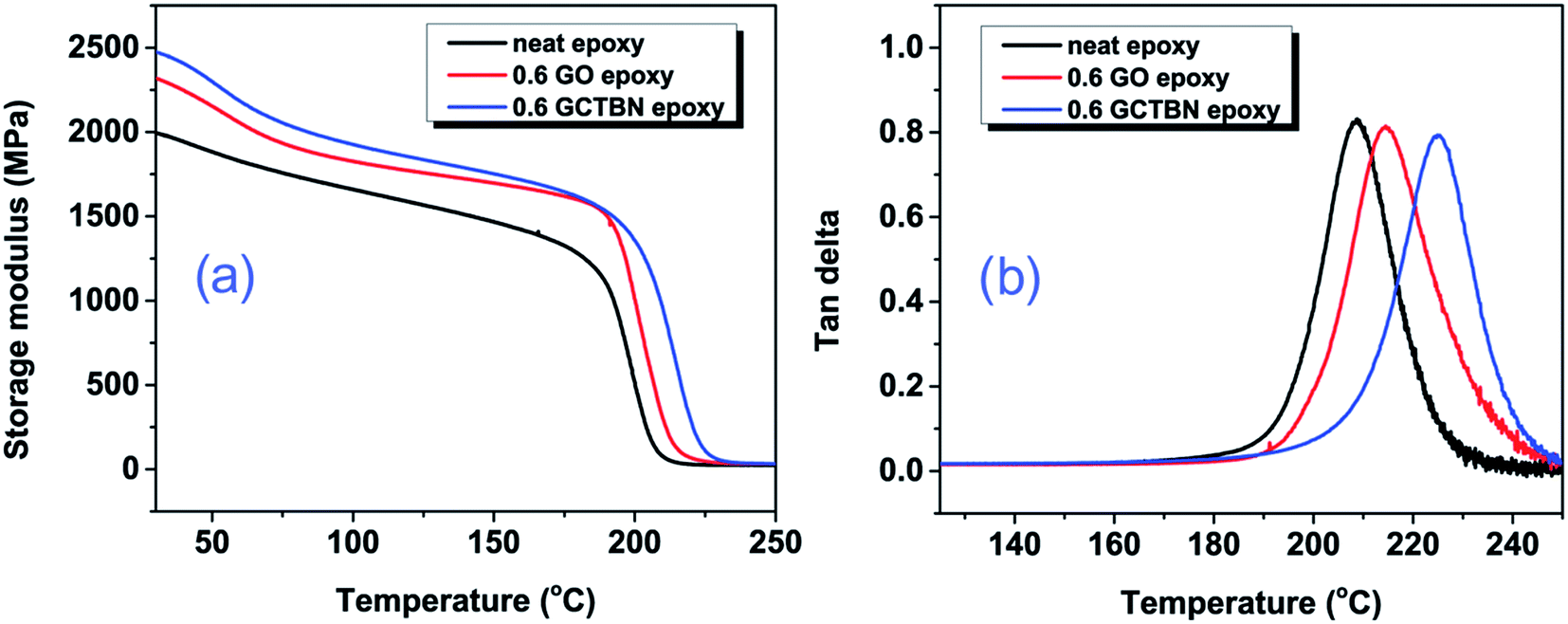 | ||
| Fig. 11 (a) Storage modulus and (b) tandelta versus temperature curves for neat epoxy, 0.6 wt% GO and 0.6 wt% GCTBN modified epoxy nanocomposite. | ||
| Sample | E′G at 30 °C (MPa) | E′R at 245 °C (MPa) | Tg (°C) | Coefficient (C) | Constrained region (Cr) |
|---|---|---|---|---|---|
| Neat epoxy | 1995 | 24.75 | 208.5 | 0 | |
| 0.6 wt% GO/epoxy | 2319 | 31.89 | 214.5 | 0.746 | 0.0040 |
| 0.6 wt% GCTBN/epoxy | 2476 | 34.72 | 225 | 0.732 | 0.0114 |
Fig. 11(b) shows the temperature dependent tanδ of cured neat epoxy and its GO composites. The tanδ is the ratio of E′′ to E′ and the peaks of tanδ are often used to determine the Tg of the material. The obtained Tg for the composites are shown in Table 2. The Tg of epoxy composites increased with the addition of GO, from 208.5 °C in the case of pristine resin to 214.5 °C for the 0.6 wt% GO/epoxy composite, with an increase of 7 °C. On the other hand, the GCTBN modified epoxy composite shows the highest Tg (∼225 °C), with a remarkable increase of 16.5 °C. This increase in Tg is due to the hindered polymer chain mobility near the filler/matrix interface or surrounding the filler due to chemical bonding.
The height depression in the tanδ peak indicates a reduction in the amount of mobile polymer chains during the glass transition; therefore the height loss in tanδ peak can be used to determine the volume fraction of the constrained region (polymer chains immobilized by the GO platelets) in epoxy phase in the epoxy nanocomposites.1,37,38 The height depression in the tanδ peak and the increase in Tg is considerably significant for the GO modified epoxy and GCTBN modified epoxy system. The volume fraction of the constrained region in each sample can be estimated from the height of the tanδ peak.38
For linear viscoelastic behavior, the relationship among the energy loss fraction of the polymer nanocomposite W and tanδ is given by the following equation.39,40
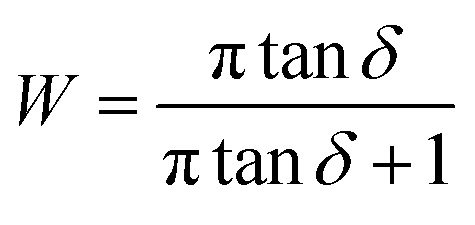 | (3) |
The energy loss fraction W at the tanδ peak is expressed by the dynamic viscoelastic data in the form.
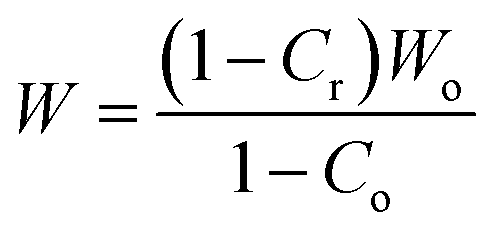 | (4) |
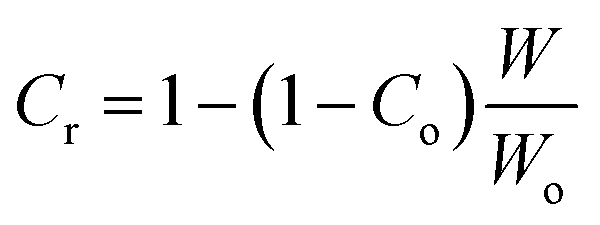 | (5) |
Co is taken to be 0 (totally amorphous phase in epoxy). The height of the tanδ peak is used to calculate W according to eqn (3). The calculated volume fraction of the constrained region is given in the Table 2.
In epoxy/GO composites, the GOs have high surface to volume ratio and hence epoxy chains get attached to the GO surface thereby limiting the mobility of the surrounding polymer chains and hence leads to the formation of constrained regions around the nano filler with higher Tg. Among the epoxy nanocomposites, the GCTBN modified epoxy system exhibits the highest fraction of constrained region. For the GCTBN modified epoxy blends, the sheet/matrix interfacial bonding is effectively improved and therefore more epoxy chains get attached with the GO surface leading to the formation of a higher fraction of constrained regions with highest Tg and height depression in the tanδ peak.
The interfacial interaction between the GCTBN and epoxy matrix can also be calculated from tanδ profile.41 The relationship between tanδ of the polymer nanocomposites and neat polymer can be evaluated by the following equation.
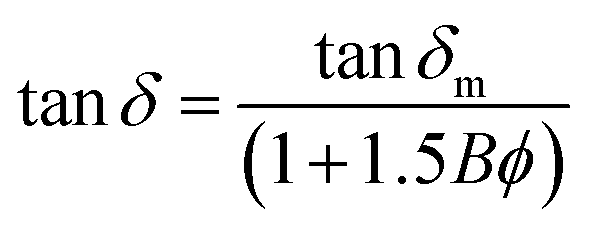 | (6) |
δ and tanδm are the loss tangent of polymer nanocomposite and neat polymer, respectively; ϕ and B represents the volume fraction of the fillers and an interaction parameter respectively. The positive value of B indicates good interaction between the fillers and polymer matrix.42
The calculated interaction parameter (B) for GO modified epoxy and GCTBN modified epoxy nanocomposites is 3.12 and 8.84 respectively. As mentioned above, the positive value of B indicates good interaction between the fillers and polymer matrix. Please note that the interaction parameter for GCTBN modified epoxy nanocomposites is much higher when compared with GO modified epoxy system. This is due to the strong interfacial interaction between the filler and matrix after chemical modification with rubber, which reduces the mobility of the local matrix around the sheets, which led to better modulus and Tg.
For further understanding of the interfacial interaction between the filler and epoxy polymer chains, the effectiveness of fillers in the composites was calculated from the storage modulus profile by using the equation.
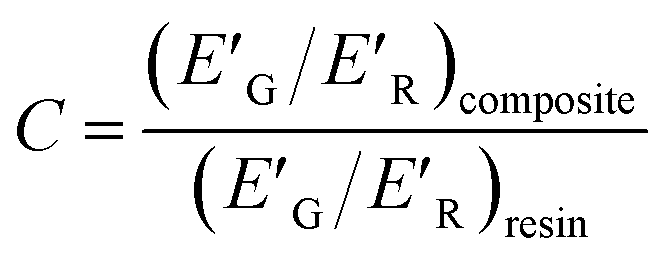 | (7) |
The lower the value of the constant C, the higher the effectiveness of the filler. The measured E′ values at 30 and 245 °C are indicated as E′G and E′R respectively. The effectiveness of the filler is found to be the highest in the GCTBN modified epoxy composites. These results are in agreement with the calculated volume fraction of the constrained region and interaction parameter.
3.6 TGA of epoxy composite
Thermal stability of the epoxy composites was traced using TGA. The thermal stability of epoxy composites with 0.6 wt% loading of GO or GCTBN were compared with neat epoxy system in Fig. 12. The thermal stability of epoxy matrix was not affected by the addition of GO. As shown in the figure, the main weight loss for the composites takes place at around 320 °C, which is attributed to the degradation of epoxy network. This means that all the composites prepared are very stable and show little degradation below 320 °C and therefore can be used for many high temperature applications.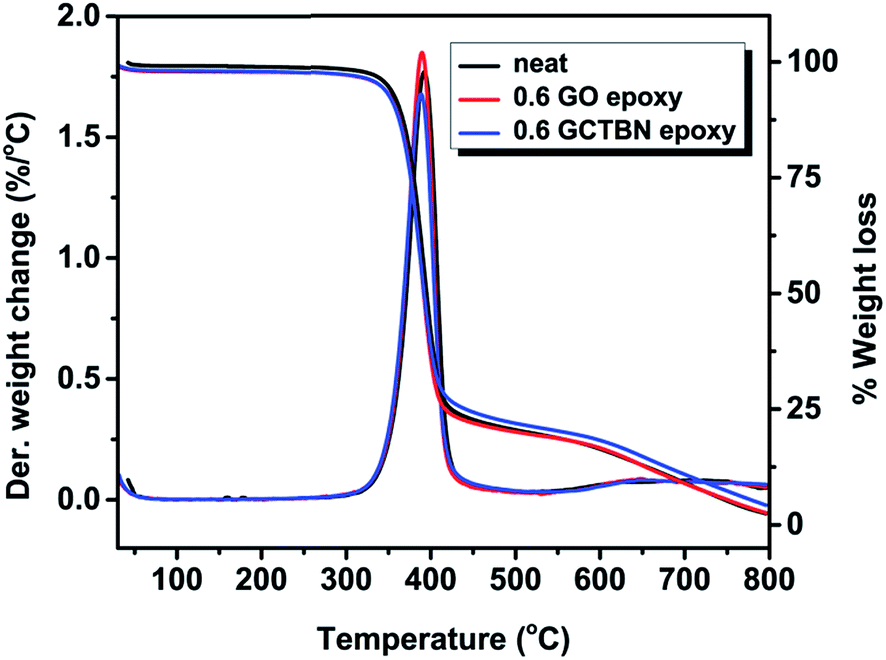 | ||
| Fig. 12 TGA and DTG curve of epoxy nanocomposites. | ||
4. Conclusion
CTBN grafted GO was successfully synthesized. Partial reduction of GO was observed during functionalisation with CTBN rubber. The prepared GO and GCTBN were characterized by FTIR, XPS, Raman spectroscopy, XRD, TEM, TOM and TGA. The studies reveals the grafting of CTBN with GO. The thermo-mechanical properties were carefully analyzed using UTM, DMA and TGA. The fracture surface was carefully characterized by SEM. The mechanical properties of the epoxy system show an excellent improvement by the addition of GCTBN. The surface morphology reveals improved interfacial bonding between the filler and the matrix. Therefore, GCTBN modified composites are able to carry higher level of loading during fracture. The viscoelastic properties also show drastic improvement in modulus and Tg. This improvement in Tg is due to the hindered polymer chain mobility near the filler/matrix interface. The improvement in modulus and Tg was further confirmed by the quantitative analysis of the volume fraction of the constrained region and interaction parameter. Moreover, the effectiveness of the filler was revealed by the value of the coefficient C. Furthermore, the TGA studies reveal good thermal stability and depicts that the decomposition temperatures of epoxy composites are above 320 °C. In addition, it can be concluded that GCTBN can be effectively utilized to improve significantly the thermo-mechanical properties of epoxy composites.Acknowledgements
R. K. wishes to hereby thank IIST for the research fellowship. The authors also acknowledge Sophisticated Analytical Instrument Facilities (SAIF), IIT Madras for the SEM analysis and Vikram Sarabhai Space Centre, Thiruvananthapuram for mechanical testing.References
- R. Konnola, J. Parameswaranpillai and K. Joseph, Polym. Compos., 2015 DOI:10.1002/pc.23390.
- R. Thomas, S. Durix, C. Sinturel, T. Omonov, S. Goossens, G. Groeninckx, P. Moldenaers and S. Thomas, Polymer, 2007, 48, 1695–1710 CrossRef CAS PubMed.
- J. H. Hodgkin, G. P. Simon and R. J. Varley, Polym. Adv. Technol., 1998, 9, 3–10 CrossRef CAS.
- J. Parameswaranpillai, P. Moldenaers and S. Thomas, RSC Adv., 2013, 3, 23967–23971 RSC.
- N. Chikhi, S. Fellahi and M. Bakar, Eur. Polym. J., 2002, 38, 251–264 CrossRef CAS.
- A. B. Leonardi, I. A. Zucchi and R. J. Williams, Eur. Polym. J., 2015, 65, 202–208 CrossRef CAS PubMed.
- P. Jyotishkumar, E. Logakis, S. M. George, J. Pionteck, L. Häussler, R. Haßler, P. Pissis and S. Thomas, J. Appl. Polym. Sci., 2013, 127, 3063–3073 CrossRef CAS PubMed.
- J. A. King, D. R. Klimek, I. Miskioglu and G. M. Odegard, J. Appl. Polym. Sci., 2013, 128, 4217–4223 CrossRef CAS PubMed.
- L.-C. Tang, Y.-J. Wan, D. Yan, Y.-B. Pei, L. Zhao, Y.-B. Li, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, Carbon, 2013, 60, 16–27 CrossRef CAS PubMed.
- J. Zhu, S. Wei, J. Ryu, M. Budhathoki, G. Liang and Z. Guo, J. Mater. Chem., 2010, 20, 4937–4948 RSC.
- A. A. Azeez, K. Y. Rhee, S. J. Park and D. Hui, Composites, Part B, 2013, 45, 308–320 CrossRef CAS PubMed.
- J. Gao, J. Li, B. C. Benicewicz, S. Zhao, H. Hillborg and L. S. Schadler, Polymers, 2012, 4, 187–210 CrossRef CAS PubMed.
- J. Parameswaranpillai, A. George, J. Pionteck and S. Thomas, J. Polym., 2013, 2013, 183463, DOI:/10.1155/2013/183463.
- R. Sengupta, M. Bhattacharya, S. Bandyopadhyay and A. K. Bhowmick, Prog. Polym. Sci., 2011, 36, 638–670 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- M. Cano, U. Khan, T. Sainsbury, A. ÓNeill, Z. Wang, I. T. McGovern, W. K. Maser, A. M. Benito and J. N. Coleman, Carbon, 2013, 52, 363–371 CrossRef CAS PubMed.
- K. Song, Y. Zhang, J. Meng, E. C. Green, N. Tajaddod, H. Li and M. L. Minus, Materials, 2013, 6, 2543–2577 CrossRef CAS PubMed.
- M. Naebe, J. Wang, A. Amini, H. Khayyam, N. Hameed, L. H. Li, Y. Chen and B. Fox, Sci. Rep., 2014, 4, 4375, DOI:10.1038/srep04375.
- Y. T. Park, Y. Qian, C. Chan, T. Suh, M. G. Nejhad, C. W. Macosko and A. Stein, Adv. Funct. Mater., 2015, 25, 575–585 CrossRef CAS PubMed.
- L.-Z. Guan, Y.-J. Wan, L.-X. Gong, D. Yan, L.-C. Tang, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, J. Mater. Chem. A, 2014, 2, 15058–15069 CAS.
- X. Wang, W. Xing, X. Feng, B. Yu, L. Song and Y. Hu, Polym. Chem., 2014, 5, 1145–1154 RSC.
- G. Tang, Z.-G. Jiang, X. Li, H.-B. Zhang, S. Hong and Z.-Z. Yu, Composites, Part B, 2014, 67, 564–570 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- G. Tripathi and D. Srivastava, Mater. Sci. Eng., 2007, 443, 262–269 CrossRef PubMed.
- H.-H. Liu, W.-W. Peng, L.-C. Hou, X.-C. Wang and X.-X. Zhang, Compos. Sci. Technol., 2013, 81, 61–68 CrossRef CAS PubMed.
- T. Jiang, T. Kuila, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2014, 2, 10557–10567 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS PubMed.
- J. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- X. Qian, L. Song, B. Yu, W. Yang, B. Wang, Y. Hu and R. K. Yuen, Chem. Eng. J., 2014, 236, 233–241 CrossRef CAS PubMed.
- F.-Y. Yuan, H.-B. Zhang, X. Li, H.-L. Ma, X.-Z. Li and Z.-Z. Yu, Carbon, 2014, 68, 653–661 CrossRef CAS PubMed.
- X. Wang, L. Song, H. Yang, W. Xing, B. Kandola and Y. Hu, J. Mater. Chem., 2012, 22, 22037–22043 RSC.
- W. Li, X.-Z. Tang, H.-B. Zhang, Z.-G. Jiang, Z.-Z. Yu, X.-S. Du and Y.-W. Mai, Carbon, 2011, 49, 4724–4730 CrossRef CAS PubMed.
- P. G. Ren, H. Wang, H. D. Huang, D. X. Yan and Z. M. Li, J. Appl. Polym. Sci., 2014, 131, 39803, DOI:10.1002/app.39803.
- J. Ma, Q. Meng, A. Michelmore, N. Kawashima, Z. Izzuddin, C. Bengtsson and H.-C. Kuan, J. Mater. Chem. A, 2013, 1, 4255–4264 CAS.
- J. Górka, R. T. Mayes, L. Baggetto, G. M. Veith and S. Dai, J. Mater. Chem. A, 2013, 1, 3016–3026 Search PubMed.
- K. Song, Y. Zhang and M. L. Minus, Macromol. Chem. Phys., 2015, 216, 1313–1320 CrossRef CAS PubMed.
- P. P. Vijayan, D. Puglia, J. M. Kenny and S. Thomas, Soft Matter, 2013, 9, 2899–2911 RSC.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, T. Kurauchi and O. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1755–1758 CrossRef CAS PubMed.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1185–1189 CrossRef CAS.
- K. D. Ziegel and A. Romanov, J. Appl. Polym. Sci., 1973, 17, 1119–1131 CrossRef CAS PubMed.
- S. Zeng, C. Reyes, J. Liu, P. A. Rodger, S. H. Wentworth and L. Sun, Polymer, 2014, 55, 6519–6528 CrossRef CAS PubMed.
- H. Varghese, S. Bhagawan and S. Thomas, J. Appl. Polym. Sci., 1999, 71, 2335–2364 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |