
Phycocyanobilin, a bioactive tetrapyrrolic compound of blue-green alga Spirulina, binds with high affinity and competes with bilirubin for binding on human serum albumin†
Abstract
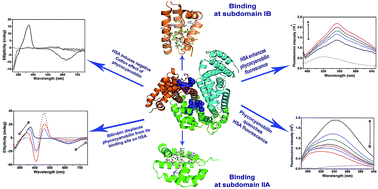
Human serum albumin (HSA) is an important regulator of the pharmacokinetic properties of bioactive compounds. Phycocyanobilin is a blue tetrapyrrole chromophore of C-phycocyanin with proven health-promoting activities. Despite its structural similarity to bilirubin, the conformation it adopts in aqueous solution is different and the pigment is more soluble than bilirubin. The aim of our study was to examine binding of phycocyanobilin for HSA and to investigate its competition with bilirubin. Based on a computational approach, we demonstrated two putative high-affinity binding pockets on HSA of virtually identical energies for the neutral and anion forms of bilirubin, but with slightly favorable predictions for anion forms of phycocyanobilin. Computational prediction of phycocyanobilin pKa values suggested a monoanion form to be the most stable form at physiological conditions. The computationally predicted binding sites for phycocyanobilin were identical to the two previously identified binding sites for bilirubin (subdomains IB and IIA). Results obtained by protein and pigment fluorescence measurements, circular dichroism, and competition experiments confirmed high affinity (binding constant of 2.2 × 106 M−1 at 25 °C), stereo-selective binding of phycocyanobilin M-conformer to HSA and its competition with bilirubin, warfarin and hemin. Our experimental data confirm that phycocyanobilin binds to IB and IIA binding site of HSA with an affinity similar to bilirubin. In conditions characterized by an increased bilirubin plasma concentration, or intake of drugs binding to IB or IIA binding site, pharmacokinetics of phycocyanobilin may also be changed.
 Please wait while we load your content...
Please wait while we load your content...

