
Methanol formation by catalytic hydrogenation of CO2 on a nitrogen doped zinc oxide surface: an evaluative study on the mechanistic pathway by density functional theory†
Ramasamy Shanmugamab,
Arunachalam Thamaraichelvana and
Balasubramanian Viswanathan*b
aDepartment of Chemistry, Thiagarajar College, Madurai, Tamilnadu 625 009, India
bNational Center for Catalysis Research, Indian Institute of Technology Madras, Chennai, Tamilnadu 600 036, India. E-mail: bvnathan@iitm.ac.in
First published on 29th June 2015
Abstract
An investigation of the nature of adsorption of H2O and CO2 on a nitrogen doped zinc oxide cluster surface and the resultant reaction between them has been performed using hybrid density functional theory (DFT) calculations at the B3LYP level of theory in a vacuum. The stable chemisorption modes of CO2 and H2O on metal, oxygen and nitrogen sites were examined. The calculated adsorption energies reveal that the formation of CO2− attached to N is the most favorable process for CO2 on the Zn18O17:N cluster surface, with a binding energy of −1.86 eV. The water molecule spontaneously dissociates on the same surface to produce chemisorbed H* and *OH with an interaction energy of −0.77 eV. The model calculations rationalize the hydrogenation of CO2 by H2 generated from H2O on the cluster surface. Thermodynamically favorable reaction pathways for the formation of methanol on the catalytic surface in a vacuum were proposed. Among the three pathways, methanol formation follows the carbamate route. The carbamate formed undergoes hydrogenation to generate COOH* units, followed by its exothermic decomposition to *CO attached to N and *OH. Further hydrogenation of CO ultimately yields methanol. All of the above steps were computationally evaluated.
1. Introduction
A rapid increase in the consumption of fossil fuels results in the emission of a huge amount of the greenhouse gas carbon dioxide into the atmosphere. The radiative forcing effect of carbon dioxide causes adverse changes in the environment.1 Partial success has been achieved in generating value added chemicals and fuels that can store renewable energies.2–9 However, the higher thermodynamic stability of CO2 makes it relatively inert towards the conversion process and thus requires activation. Among the metal oxide semiconductors, ZnO exhibits a greater potential in activating inert CO2 into active CO2− species on its surface.10 The interaction of CO2 and H2O on the ZnO surface has been studied extensively using DFT methods.11–14 Although the formation of carbonate and hydrogenated carbonate species on the surface has been confirmed, the fact that they restrict further activation has also been proven by in situ techniques.15Industrially, Cu/ZnO/Al2O3 was used as a catalyst to synthesize methanol from a mixture of H2, CO2 and CO, which requires high temperature and pressure.16 On ZnO, the photoreduction of CO2 and H2O yields methane as the major product,17 in contrast to the high temperature methanol synthesis on the catalytic surfaces. In sharp contrast, the enzyme carbonic anhydrase activates and fixes carbon dioxide as bicarbonate in nature, surprisingly at room temperature. It is better to produce chemicals rather than simply mimicking photosynthesis. Hence, based on the photosynthetic enzyme model, various approaches were tried to use zinc complexes for CO2 activation.18–21 For solar energy conversion, semiconductor-based photocatalytic systems have been employed in numerous applications, which opened up a new era for the utilization of 43% of the available solar radiation in the visible range for hydrogen production from water, as well as for CO2 reduction.8,9,17,22–26 In recent years, Ni supported on SiO2–Al2O3 has been used to produce methane using CO2 and H2 under solar energy conditions,27 while ZnO-coated CuO yielded CO.28 ZnO based catalysts have been shown to participate in the water splitting reaction too.29–31 Furthermore, since ZnO is active in the UV region, it undergoes photocorrosion during these reactions. For the efficient reduction of CO2, protons and electrons need to be continuously supplied during the reaction.
Nitrogen-containing catalytic systems have recently received greater attention to overcome the above difficulty, due to their greater potential to adsorb as well as activate carbon dioxide.32–38 Nitrogen doped zinc oxide plays a vital role in catalysis, sensors, and optoelectronic devices.39,40 Further, N doped ZnO performs better than undoped catalysts in photocatalytic water splitting in the visible region.41,42 The activation of CO2 mainly concerns the addition of an electron, that alters the bond angle from linear 180° to a near tetrahedral or triangular angle, for the formation of methanol and carbonate-like products respectively. The N-containing molecules in the homogeneous catalytic systems lead to an O–C–O angle of 132° in the adjacent oxygen environment.19 Hence, these kind of surfaces could provide a better environment for CO2 activation.
The interactions of H2O and CO2 on various photocatalytic material surfaces have been extensively studied.11,13,14,43–51 The reduction of CO2 by H2O is controlled by the surface configuration and adsorption strength of the adsorbate on the active sites of a catalyst. The nature of the interactions of CO2 and H2O on a nitrogen doped zinc oxide surface has not yet been studied. Further, understanding of the basic molecular level interaction mechanism can provide new insight into the design of a better catalyst. Hence, in the present study, a systematic investigation of the adsorption and activation of CO2 and H2O on a nitrogen substituted stoichiometric ZnO nanocluster (Zn18O17:N) by means of a DFT method is reported. Furthermore, the plausible reduction mechanism of CO2 to methanol has also been proposed. We expect that this fundamental study would help to understand and identify the molecular events taking place on specific surfaces that are required to design newer and efficient catalysts for future reactions.
2. Molecular models and methods of computation
2.1. Creation of a N doped stoichiometric Zn18O17:N cluster
Bulk ZnO crystallises in a wurtzite structure. The surface was terminated with either oxygen or zinc sites, with four fold coordination dominating over the three or two fold analogues. Since surface relaxation alters the surface termination at the nano level, both kinds of termination are possible on the surface.52 For the Zn12O12 cluster, the embedded ZnO (000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) was used to study the interaction of CO2 on the surface,13 which has many vacancies. Hence, to minimize the vacancies at the cluster level, Zn18O18 was chosen as the initial structure in order to account for the stoichiometry, wurtzite configuration and the occurrence of Zn and O atoms in different coordination environments. This geometry was optimized and the resultant configuration is presented in Fig. 1.
) was used to study the interaction of CO2 on the surface,13 which has many vacancies. Hence, to minimize the vacancies at the cluster level, Zn18O18 was chosen as the initial structure in order to account for the stoichiometry, wurtzite configuration and the occurrence of Zn and O atoms in different coordination environments. This geometry was optimized and the resultant configuration is presented in Fig. 1.
 | ||
| Fig. 1 Schematic representation of the optimized Zn18O18 cluster. | ||
One of the four fold sites of O was replaced by N to generate the stoichiometric Zn18O17:N, considered as a N doped ZnO model surface for computational studies. The introduction of N on the ZnO surface creates the heterogeneity of atoms. Suitable modes of adsorption on metal oxides and nitrides were proposed for H2O and CO2.44,46–53 However, no such modes of adsorption are available for the corresponding nitrogen doped surface. Hence, new modes of adsorption on the Zn18O17:N surface were proposed for H2O and CO2, combining those previously reported. Modes such as (a) and (f) on top monodentate linear (CO2, η1-O), (b) and (g) bent monodentate (CO2, η1-C), (c) and (i) bridge bidentate (CO2, η2-C,O), (d) and (h) bridge tridentate (CO2, η3-O,C,O), and finally, (e) and (j) bridge bidentate (CO2, η2-O,O) have been considered as adsorption models for CO2. For water, (k) and (o) on top monodentate (H2O, η1-O); (l) on top monodentate (H2O, η1-H), (m), (n) and (p) bridge bidentate (H2O, η2-O,H) and finally (q) bridge bidentate (H2O, η2-H,H) modes of adsorption were proposed as models, which are depicted in Fig. 2.
 | ||
| Fig. 2 Schematic representation of the various adsorption modes for CO2 [(a)–(j)] and for H2O [(k)–(q)] on a stoichiometric Zn18O17:N cluster. | ||
The surface reconstruction during the adsorption process could provide more information on the surface reaction. For this reason, full relaxation of the adsorbates and cluster was allowed through geometry optimization. For clarity, the surface atoms with differing four, three and two fold coordinative sites are designated as 4fc, 3fc and 2fc respectively. In the adsorbed state, similar atoms in CO2 and H2O are labeled as Oa, Ob and Ow, and Ha and Hb respectively in the results and discussion section.
2.2. Computational details
Geometry optimization and all other electronic calculations were computed employing Density Functional Theory (DFT) coupled with the gradient-corrected B3LYP functional method. The electrons present in the Zn atom were treated with an effective core potential of LANL2DZ.54 For other atoms, the aug-cc-pvdz55 basis set was employed. Vibrational frequency calculations were carried out to identify and locate the intermediates corresponding to minima in the potential energy surface that do not have any imaginary/negative vibrational frequency. The extent of the interaction between the adsorbate and the cluster was evaluated through the calculation of the interaction energy,56 Eads = E(adsorbate+cluster) − (Eadsorbate + Ecluster), where E(adsorbate+cluster), Eadsorbate and Ecluster are the Zero Point Energy (ZPE) corrected total energies of the adsorbate + cluster, bare adsorbate and cluster, respectively.The Gibbs free energy for a given reaction step was calculated using the relation, ΔGo = ∑Goproducts − ∑Goreactants, where Goreactants and Goproducts correspond to the ZPE corrected standard free energies (without any scaling factor) of the reactants and products respectively at the temperature of 298.15 K and the pressure of 1 atm.
The ab initio molecular dynamics (AIMD) computation was performed using an extended Lagrangian Atom Centered Density Matrix Propagation Molecular Dynamics (ADMPMD) at the B3LYP/LANL2DZ level to check the stability of the adsorbate on the surface. The final structure obtained from the optimized geometries of the adsorbate with Zn18O17:N was chosen as the initial geometry for the ADMPMD run. A dynamics simulation was carried out using 1000 steps with the time scale of 0.1 femtosecond and the fictitious electron mass of 0.1 amu. The thermostat temperature was maintained at 300 K using a velocity scaling method.57
Natural Bond Orbital (NBO) analysis58 was used to characterize the charge transfer on the surface. All of the electronic properties were computed using the Gaussian 09 software package.59 The density of states was analysed using the spin polarized GGA-PBE method, with an energy cut off of 380 eV and auto generated k points in CASTEP code implemented in Material Studio 5.5.60
3. Results and discussion
3.1. Zn18O17:N geometries and the chemical state of doped N
For the nitrogen doped stoichiometric Zn18O17:N cluster, geometry optimization was performed using hybrid density functional theory. After finding the global minima configuration, the structural parameters were analyzed. The optimized configuration for Zn18O17:N is shown in Fig. 3(a). Upon doping of ZnO with N, the resulting cluster experienced modified structural parameters. Out of the four Zn–N bonds, where N has 4fc, three are present on the same surface and have a bond distance of 2.04 Å. However, the fourth one was elongated to a distance of 3.50 Å, thus indicating the absence of a fourth bond. The presence of nitrogen in a three fold coordinative site is favored and causes a slight expansion of the crystal. This effect was experimentally observed in ZnO doped with N, leading to a small shift in the 2θ value.61 The bond length of Zn–O lies in the range of 1.93 to 1.99 Å, the variation arising due to the surface relaxation of the atoms. Furthermore, the oxidation state of the surface N was evaluated from the Partial Density of States (PDOS) of Zn18O17:N as shown in Fig. 4. The overall DOS population of Zn18O17:N revealed a population profile similar to that observed in Zn18O18, thus confirming the incorporation of N on the surface. The N incorporated into ZnO exists as N3−, which is isoelectronic with the O2− ion. This attributes an excess of electron density on the surface N in relation to the oxide species, which is confirmed from the spin density analysis of Zn18O17:N, as depicted in Fig. 3(b). | ||
| Fig. 3 Optimized structure of (a) Zn18O17:N and (b) spin density distribution in Zn18O17:N. | ||
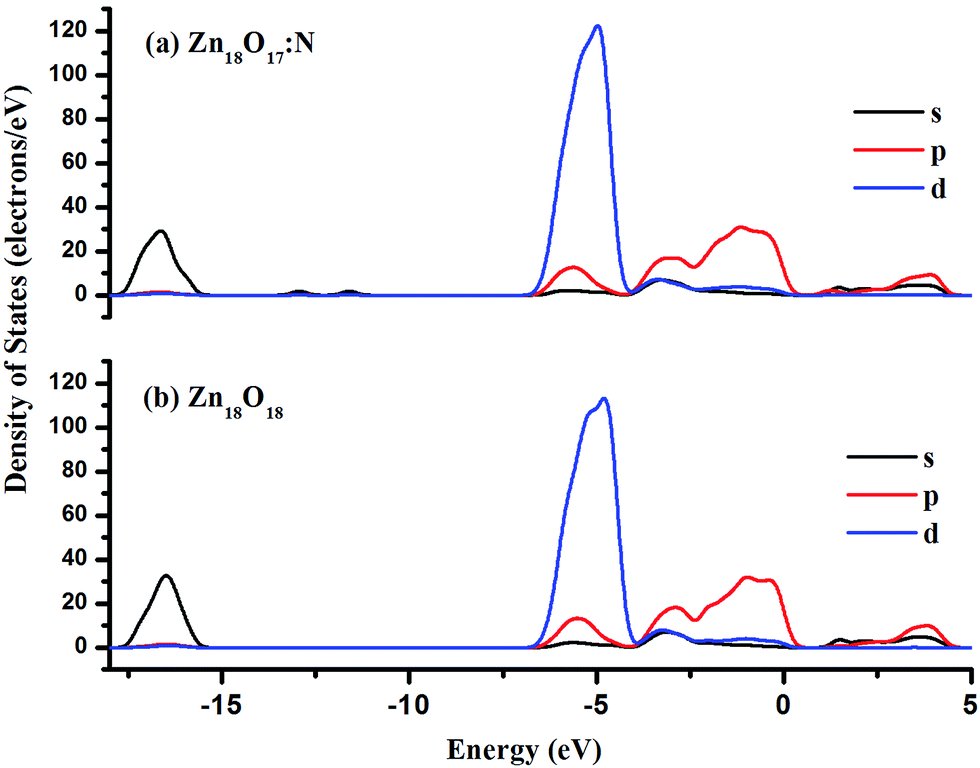 | ||
| Fig. 4 Partial density of states population of (a) nitrogen doped zinc oxide and (b) pure ZnO clusters. | ||
3.2. CO2 adsorption on Zn18O17:N
The adsorption mode of CO2 on a nitrogen doped zinc oxide surface has not been known yet. Hence, ten different molecularly adsorbed predetermined configurations of CO2 were chosen and relaxed. After finding the global minimum, the physisorption or chemisorption was characterized by calculating the adsorption energies. The structural parameters and adsorption energies for the selected configurations are presented in Table 1. For the adsorption of CO2 at zinc sites with varying coordinative environments on the Zn18O17:N surface, the adsorption energies were computed using both the CO2,η1-O and CO2,η1-C monodentate linear vertical to the surface modes. After relaxation, both the geometries yielded the same configuration as shown in Fig. 5(a).| Configuration | ΔE | d | l(C–Oa) | l(C–Ob) | θ | q | |||
|---|---|---|---|---|---|---|---|---|---|
| N | C | Oa | Ob | ||||||
| CO2 | — | — | 1.16 | 1.16 | 179.9 | — | 1.069 | −0.535 | −0.535 |
| a | −0.48 | 2.5 | 1.16 | 1.17 | 176.8 | −1.455 | 1.115 | −0.576 | −0.506 |
| b | −1.14 | 1.33 | 1.27 | 1.27 | 122.9 | −1.472 | 1.081 | −0.833 | −0.803 |
| c | −1.86 | 1.35 | 1.28 | 1.29 | 123.7 | −0.840 | 0.867 | −0.797 | −0.808 |
 | ||
| Fig. 5 Optimized structures for the various coordinative adsorption modes of CO2 leading to (a) physisorption, (b) a bridge tridentate carbonate and (c) a bridge tridentate carbamate on Zn18O17:N. | ||
The binding energies for the adsorption of CO2 on metal sites in pure metal oxides have been shown to follow the weak Eley–Rideal physisorption model, rather than chemisorption. The CO2 was held on the surface without effective charge transfer from the surface. The structural parameters of CO2 were not affected62 and were indicative of free CO2 in the gas phase (Table 1). Thus, CO2 does not get activated on metal sites in the pure metal oxide frame work. However, when the interaction of the bidentate and tridentate modes of CO2 on the coordinatively unsaturated sites of oxygen, zinc and nitrogen of Zn18O17:N were considered, chemisorption occurs. The resultant structures are presented in Fig. 5(b and c). The adsorption of CO2 (η1-C) at the three fold coordination site, O(3fc), on the edge of the surface leads to the carbonate formation via the two oxygens coordinating with the two adjacent Zn(3fc) sites, thus saturating the surface oxygen vacancy48 (Fig. 5(b)). The angle of the O–C–O bond was altered to 122.9° from 180°, which is closer to triangular. The C–Oa and C–Ob bond lengths were elongated from 1.16 to 1.27 Å with the binding energy of −1.14 eV. These values correlate well with the tridentate carbonate structure on the ZnO surface.48 In addition to the evaluation of the charge donor/acceptor interactions, NBO charge analysis indicates that the charges on C, Oa and Ob acquired an excess value of −0.555e, indicating the effective interaction with the surface. The calculated vibrational frequencies have real values, indicating the stable nature of the structures. The O–C–O asymmetric stretching mode occurs at 1580 cm−1. The surface adsorbed CO2 reveals the Osurface–C stretching mode at 1348 cm−1, while the carbonate-like three C–O units stretch at 1045 cm−1. The decrease in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching is due to the loosening of the bond as a result of the adsorption through only one oxygen atom. The larger decrease in frequency in the carbonate structure is experienced due to a decrease in the bond order in the two coordinating CO groups. The out-of-plane bending mode of CO2 occurs at 846 cm−1. All calculated frequencies are consistent with the reported experimental values15 and suggest that even if the surface has an impurity of N, the adsorption nature of the oxide does not alter.
O stretching is due to the loosening of the bond as a result of the adsorption through only one oxygen atom. The larger decrease in frequency in the carbonate structure is experienced due to a decrease in the bond order in the two coordinating CO groups. The out-of-plane bending mode of CO2 occurs at 846 cm−1. All calculated frequencies are consistent with the reported experimental values15 and suggest that even if the surface has an impurity of N, the adsorption nature of the oxide does not alter.
The binding energy of CO2 in η3 adsorption on N(4fc) is −1.86 eV, with a chemisorption distance of 1.35 Å for N(4fc)–C. After the insertion of CO2, the bonds surrounding N are broken. The upward movement of the N atom reveals its dangling nature, which is enhanced by greater adsorption. The two coordinatively unsaturated Zn atoms in the vicinity are also pushed upward to facilitate coordination with the oxygen atoms, with the distances of Oa–Zn and Ob–Zn being 2.03 Å and 2.04 Å respectively (Fig. 5(c)). Further, the adsorption energy of the same mode on pure Zn18O18 was calculated (ESI 1†) to examine the effect of nitrogen. For the pure Zn18O18 clusters, the value is −1.06 eV, which is lower than that for the N substituted surface by −0.80 eV. This result indicates that the presence of nitrogen enhances the adsorption.
The comparison of the adsorption energies reveals that the tridentate carbamate species is more favorable than the carbonate species. It may be due to the greater electron density on N and its lower electronegativity than O, which favors effective charge transfer. The Gibbs free energy profile is depicted in Fig. 6 for the above process. First, CO2 interacts with the surface by physisorption (Fig. 6, S1). Then the C interacts with the N, leading to distortion of the O–C–O angle (Fig. 6, S2). The resultant bent configuration of the bidentate species has a relative Gibbs free energy of −0.77 eV. The values of the calculated vibrational frequencies show that this species is a transition state, which undergoes further changes to form a stable tridentate species (Fig. 6, S3).
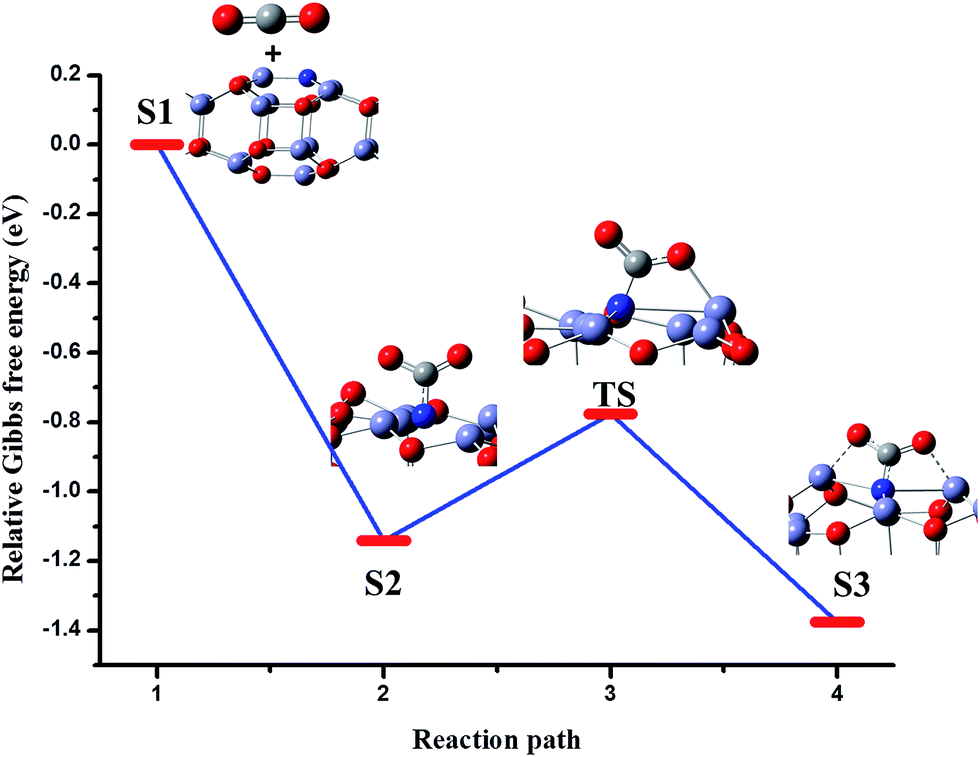 | ||
| Fig. 6 Relative Gibbs free energy profile with the corresponding structures for the reaction of CO2 to CO2− on the surface of stoichiometric Zn18O17:N to form N-carboxylate. | ||
The excess charge on CO2 is −0.738e, which is greater than that acquired on tridentate carbonate (−0.555e). This charge transfer from the surface reveals that the N doped zinc oxide is a more powerful Lewis base than pure zinc oxide. In addition, there is no imaginary frequency value in the vibrational spectrum, that indicates its stable nature. The Oa–C–Ob asymmetric stretching mode occurs at 1461 cm−1. The surface N–C stretching, and the symmetric stretching that combines C–O and N–C are observed at 1366 cm−1 and 1063 cm−1 respectively. At 823 cm−1, the out-of-plane bending of C in the carboxylate is revealed. These patterns have also been observed in organic carbamate species.63
Further investigation of the carbamate stability on the surface was done using ADMPMD calculations. The distance between the carbamate and the surface at 300 K is shown in ESI Fig. 1(a).† The plot reveals that during the dynamical run, the adsorbed CO2 is held on to the surface for up to 1 ps without any desorption.
The density of states analysis provides information such as where exactly the surface transferred electrons are populated on the adsorbate. Thus, the partial density of states (PDOS) for the s and p orbitals of CO2 corresponding to free CO2, physisorbed linear CO2, tridentate carbonate and tridentate carbamate were plotted (Fig. 7). The PDOS population of CO2 in the physisorbed state reveals that there is no effective interaction with the surface. For the tridentate carbonate species, the p orbital is more populated in the range of −7.5 eV to 0 eV at the Fermi level. This indicates that the surface effectively transfers the charge to the adsorbate in the activated form. Although the tridentate carbamate has the same pattern, the population is higher than that of carbonate. Thus, activation in the presence of nitrogen is more facile than in a pure oxide environment.
 | ||
| Fig. 7 Partial density of states population of (a) free CO2, (b) physisorbed linear CO2, (c) tridentate carbonate and (d) tridentate carbamate species on nitrogen doped zinc oxide clusters. | ||
3.3. Adsorption of H2O on Zn18O17:N
The presence of N produces a number of possible adsorption mode configurations suitable as active sites for H2O on the Zn18O17:N surface. To find the favorable sites for preliminary H2O adsorption, numerous adsorption sites were inspected. As sites other than N behave similar to those of the pure metal oxide, the atoms present around N in the surface have been considered as adsorption models for initial H2O adsorption. On optimization, only three of them yielded thermodynamically favorable sites as shown in Fig. 8. Table 2 summarizes the structural parameters. | ||
| Fig. 8 Optimized structures of H2O adsorbed on Zn18O17:N; (a) & (c) dissociative and (b) molecular adsorption. | ||
| Configuration | ΔE | l(H–Oa) | l(H–Ob) | θ | q | |||
|---|---|---|---|---|---|---|---|---|
| N | O | Ha | Hb | |||||
| H2O | — | 0.96 | 0.96 | 104.7 | −0.958 | 0.479 | 0.479 | |
| a | 0.02 | — | 0.96 | — | −1.237 | −1.230 | 0.401 | 0.478 |
| b | −0.77 | 0.96 | 0.99 | 107.7 | −1.451 | −0.999 | 0.498 | 0.535 |
| c | −0.44 | 0.96 | — | 108.5 | −1.495 | −1.006 | 0.515 | 0.515 |
Using the η2 (a) mode, adsorption leads to the autodissociation of H2O, with a binding energy of 0.02 eV. This value suggests that the dissociation on the N active site is slightly endothermic in nature. The Ha gets adsorbed on the surface N, and the O–Hb on the adjacent Zn atom, with adsorption distances of 1.06 Å and 1.96 Å respectively. This correlates well with the dissociation of H2O on a Ta3N5 (100) surface.46 Now, the upwardly elongated N in the three fold coordinative site contracted to a distance of 2.37 Å from 3.50 Å, with respect to the bottom layer. The bond length of O–Hb was found to be 0.96 Å, which is similar to the bare free O–H distance in H2O. The NBO charge analysis (Table 2) shows that the charge on N decreases from −1.469e to −1.237e, while the charge on O increased from −0.958e to −1.230e, indicating that the excess charge has been transferred from the surface, which causes the dissociation of a H atom.
IR frequencies revealed that there is no negative vibration in the frequencies, indicating that the configuration is a minimum. The vibrational frequency values corresponding to O–Ha symmetric stretching at 3836 cm−1, N–Hb symmetric stretching at 2885 cm−1, (N–Hb + O–Ha) in-plane wagging (1014 cm−1), (N–Hb + O–Ha) out-of-plane wagging (1014 cm−1) and the O–Ha bent vibration (737 cm−1) support the above observation.
Another favorable mode of adsorption of H2O is using the coordination of both O and H atoms to the three fold coordinated Zn and O sites respectively in a vertical fashion, as shown in Fig. 8(b). The Zn atom, present adjacent to the four fold coordinated Zn, was slightly displaced upward from the top layer and was held at a distance of 2.15 Å. The calculated binding energy of H2O in the adsorbed state was −0.77 eV. The same kind of adsorption occurs for H2O on a Zn2GeO4 surface.44 The bond lengths of O–Ha and O–Hb are 0.96 and 0.99 Å respectively. The bond distances of 2.15 Å for Znsurface–Owater and 1.70 Å for Osurface–Hwater indicate weak adsorption. NBO charge analysis predicted the charge on the individual O as −0.999e, on Ha as 0.498e and on Hb as 0.535e, revealing that there is no effective charge transfer from the surface to the adsorbate. The vibrational frequency analysis predicted that all the configurations were stable. The various frequencies calculated are: free O–Hb symmetric stretching (3841 cm−1), adsorbed O–Hb symmetric stretching (2984 cm−1), O–Ha bending (1583 cm−1) and O–Hb adsorbed bending (563 cm−1).
The horizontal adsorption of H2O on ZnO at the corner site leads to autodissociation. The O–Ha was attached to Zn(3fc) and the Hb was attached to the adjacent O(3fc) with an adsorption energy of −0.44 eV, as shown in Fig. 8(c). This value is relatively higher than that of (a). The bond lengths are O–Ha = 0.96 Å and Osurface–Hb = 1.00 Å. The vibrational frequencies suggest the existence of free Osurface–Hb, with a value of 3069 cm−1. The stretching vibrations of the chemisorbed O–Ha are observed at higher values than those for Osurface–Hb at 3851 cm−1 and O–Hb out-of-plane bending at 1020 cm−1, all being positive.
In order to confirm the stability of the dissociatively adsorbed H2O on the surface, an ADMPMD run was computed, the results of which are presented in ESI Fig. 1(b).† The plot reveals that the dissociated species were present on the surface for up to 1 ps without desorption, indicating the stability of the dissociated species on the surface.
To further evaluate the interaction of the catalytic surface with H2O, the partial density of states for various configurations were investigated. Fig. 9(a)–(d) represent free H2O, dissociative adsorption at the N site, molecular adsorption at the metal–oxygen site and dissociative adsorption at the metal–oxygen site respectively. For dissociative adsorption, the PDOS of (b) resembles that of (d), which indicates that the dissociation of water on both sites is facilitated. Furthermore, the population in (b) around the Fermi level is small. This helps effective electron charge transfer to the p orbital from the surface N. The molecularly adsorbed configuration (c) has an overlapping of orbitals that are less populated than the dissociated species, revealing that the molecular nature still exists on the surface. Preceding the stage of hydrogenation, the source of hydrogen has to be predicted. The adsorption energies reveal that CO2 gets competitively adsorbed on the N active site on Zn18O17:N. The H2O adsorption and dissociation is preferred on the N site, as well as the oxide site. However, water was preferentially dissociating on the corner site of the cluster, similar to the dissociation of water on the pure oxide surface. Hence, the source of hydrogen was assumed to be from the vacant corner site.
 | ||
| Fig. 9 Partial density of states population of (a) free H2O, (b) & (d) dissociative and (c) molecular adsorption. | ||
3.4. Co-adsorption of CO2 and H2O
H2O hinders the adsorption of CO2 by competing with it and leads to poisoning of the adsorbent. On the other hand, H2O was used as a reactant, giving rise to the bicarbonate ion. The results obtained from the single molecule adsorption calculations on the co-adsorption of CO2 and H2O were employed to verify the above fact by looking for any significant interaction on the surface. The bimolecular adsorption of water and carbon dioxide was carried out by choosing the stable configuration for the catalyst, placing one carbon dioxide adsorbed through carbon on the surface N, and water in the side-on adsorption mode as initial configurations. The structural features of two modes (Fig. 10) of co-adsorbed configurations surrounding the N substituted environment are presented in Table 3. The calculated binding energy shows that the carbamate configuration is more favorable in the co-adsorption mode. In Fig. 10(a) and (b), the water molecule coordinates in a bidentate fashion through H to O in (a) and N in (b), and O to the Zn. | ||
| Fig. 10 Optimized structures for the co-adsorption of CO2 and H2O on Zn18O17:N (a) & (b). | ||
| Model | ΔE | l(O–Ha) | l(O–Hb) | θ | l(C–Oa) | l(C–Ob) | θ | q | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O | Ha | Hb | C | Oa | Ob | ||||||||
| a | −2.27 | 0.99 | 0.96 | 107.7 | 1.28 | 1.28 | 122.6 | −1.001 | 0.536 | 0.513 | 0.868 | −0.813 | −0.807 |
| b | −2.15 | 0.97 | 0.96 | 106.1 | 1.29 | 1.28 | 122.4 | −0.971 | 0.525 | 0.511 | 0.875 | −0.812 | −0.835 |
The final configurations indicate that the η1-C of CO2 in (a) and (b) have bent structures, and the water is in the side-on adsorption mode with total binding energies of −2.27 eV and −2.15 eV respectively. Both molecules have retained their original configurations in (a) and (b) even though the adsorption modes are different, suggesting that the adsorption of CO2 on N would facilitate activation in the free as well as in the combined forms. The structural features of the water molecules are not altered in all the configurations. The CO2 angle was altered to 122°, which is lower by 5° than in the carbamate species. Thus, in the presence of a water molecule, the adsorption and activation of CO2 are facilitated more via hydrogen bonding-like interactions that favor the adsorption.
After locating the global minima (a) configuration in (Fig. 10), the further stability and interaction of the species were evaluated using the ADMPMD run method. The calculated ADMPMD profile is depicted in ESI Fig. 1(c).† The plot shows that the co-adsorbed H2O and CO2 on the surface are retained, without migration from their adsorption site, till 1 ps. This reveals that the surface water does not affect the chemisorbed CO2 by forming bicarbonate related species.15 Hence, in the mechanistic pathway studies, the water molecules were not included, as it was assumed that the water molecules have no effect.
3.5. Hydrogenation of CO2
The hydrogenation of CO2 to methanol was processed with various complex transition states and intermediates. On the metal oxide surface, the formation of carbonate, carboxylate and formate species have been proposed as intermediates for CO2 reduction reactions, depending on the reaction environment.64,65 Detailed reduction mechanisms are available for pure metals66,67 and metal oxides.47,67,68 However, for N substituted metal oxides, not enough data are available to understand the product formation. Hence, understanding the principal process involved in the hydrogenation of CO2 by means of a DFT mechanistic pathway would predict the favorable configurations and give insight into the stability of the products and transition states.Since the active site for CO2 reduction is N, the hydrogen would interact with the adjacent Zn and O sites on the surface. On these sites, H2 heterolytic dissociation was favorable, and H easily migrated due to the action of spillover.69–71 The surface was covered with hydrogen and the effect of surface hydrogen on the reaction was negligible.72 The hydrogenation of CO2 by H2 on a ZnO surface has been reported.51 Based on the above results, the following reaction pathway was proposed and tested for methanol formation. The schematic representation of the mechanistic pathway is shown in Fig. 11. The relative Gibbs free energy profile of the hydrogenation of CO2 to methanol is depicted in Fig. 12. During the catalytic cycle, the reactant CO2 was adsorbed on the catalytic surface and activated by chemisorption on N as the CO2− species.
 | ||
| Fig. 11 Reaction path for the reaction of CO2 with H2 to form CH3OH on the Zn18O17:N surface. | ||
 | ||
| Fig. 12 Relative Gibbs free energy profile for the reaction of CO2 with H2 to form CH3OH on the Zn18O17:N surface. | ||
The next step in the catalytic cycle is that the surface bound (S1) activated *COO− abstracts a proton onto the carbamate oxygen, forming an O–H bond.64 As a result, the carboxylic acid group of η2-COOH* is present perpendicular to the surface, as shown in Fig. 11 (S2). The relative Gibbs free energy was found to be −2.55 eV, indicating the spontaneous nature of the process with respect to free CO2 and H2. In another possibility based on the proton coupled electron transfer, the H was added to the C to form a formate species. But the added H was found to instantly migrate to N. This indicates that the carbon valency was fully saturated by the surface bond. These results reveal that the proton would be easily added to the carbamate species, and the formation of a formate species was ruled out on the carbamate. The Ob–H was far away from the surface in S2, and the carbonyl has the interaction with the nearby Zn atom which is slightly moved above the surface. The Oa–C–Ob angle of 117° suggests that the angle was stretched compared to the S1 state. As per the proposed mechanism, there is a possibility of either dissociation of the Oa–H in S2 leading directly to S4, or the addition of H onto Ob to form a diol (S3) species. The diol further eliminates H2O to form S4. However, the relative free energy profile indicates that the COOH*(S2) capturing a proton (at the OH moiety) and releasing water is a more favorable route than the other one. This is due to the fact that compared to the C–Oa bond (1.27 Å), the C–Ob bond (1.37 Å) is fairly weakened and favors dissociation.
*CO remains chemisorbed on the surface (S4) with the relative Gibbs free energy of −2.40 eV, which is more positive compared to S2. The Nsurface–C–Oa angle was found to be 176.38°, that makes it nearly parallel to the surface. The Oa was bonded to the adjacent Zn(3fc) atom in Nsurface–C–Oa, revealing that surface coordinative unsaturation is required for the stabilization of the surface species. Better reduction of CO2 requires the formation of a CO species from *COOH that should be spontaneous in nature.73 On the oxide surface, the formation of CO was difficult.64 However, it is favored by N substitution in the ZnO surface. Hence, the chemisorbed CO* alone is present on the surface (S4).
Further attack of hydrogen on carbon is more favorable and yields a chemisorbed formyl species (S5) with the relative Gibbs free energy of −2.59 eV, indicating the spontaneity of the reaction. The H–C–O angle is 115.6°, and the C–H and C–O distances are 1.11 Å and 1.27 Å respectively. Further attack of H leads to the formation of a H2CO* species (S6) on the surface with the relative Gibbs free energy of −1.39 eV. This process is slightly exothermic in nature. Now, the hydride species present in the medium will react with oxygen to form methylene alcohol (S7) with a relative Gibbs free energy of −1.60 eV via a spontaneous and endothermic route. The attack of H on carbon in S7 leads to the final product, methanol. Once the methanol is formed, the valences of the atoms are satisfied and during the optimization, they get relieved from the surface but are held on to the surface by a weak physical interaction (S8). On removal of methanol from the surface, the active site is regenerated and is ready for further uptake of CO2.
4. Conclusion
DFT calculations were carried out for the first time to explore the adsorption and activation of CO2 and H2O and the reduction mechanism of CO2 to methanol by hydrogenation on Zn18O17:N model surfaces. The calculations were performed at B3LYP with all the plausible modes of adsorption. The most favorable mode of adsorption for CO2 is through mono chemisorption via carbon on N, and an electrostatic interaction with the neighboring oxygen atoms. The present study reveals that the CO2 is activated to a greater extent with a bond angle of 127° in carbamate and 122° in the co-adsorption of H2O and CO2. In the case of H2O, dissociative adsorption was favored. The presence of N enhanced the adsorption energies in relation to pure Zn18O18. The hydrogenation of CO2 to CH3OH proceeds through carbamate, carbon monoxide, formyl, formaldehyde and methylene alcohol species. The results indicate that the formation of CO via the dissociation of carbamate, rather than the diol route, required a barrier of −2.48 eV. Since all the reaction steps were associated with negative free energies, the methanol formation was achieved at lower cost than the conventional methods. Further studies on N-containing defective surfaces may lead to more insight into methanol formation.Acknowledgements
We acknowledge the Department of Science and Technology (DST), India, for setting up the National Centre for Catalysis Research (NCCR). The authors also acknowledge the High Performance Computing Environment (HPCE), Indian Institute of Technology Madras (IITM) for providing computational facilities. The author R. S. is thankful to the Council for Scientific Industrial Research (CSIR) for providing financial support, vide Ref. No. 08/117(0001)-2013-EMR-I.References
- P. Falkowski, Science, 2000, 290, 291–296 CrossRef CAS PubMed.
- S. Toshiyasu, C. Jun-Chul and Y. Hiroyuki, Chem. Rev., 2007, 107, 2365–2387 CrossRef PubMed.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- R. J. Pearson, M. D. Eisaman, J. W. G. Turner, P. P. Edwards, J. Zheng, V. L. Kuznetsov, K. A. Littau, L. di Marco and S. R. G. Taylor, Proc. IEEE, 2012, 100, 440–460 CrossRef CAS.
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- C. R. Somnath, K. V. Oomman, P. Maggie and A. G. Craig, ACS Nano, 2010, 4, 1259–1278 CrossRef PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- J. B. L. Martins, V. Moliner, J. Andrés, E. Longo and C. A. Taft, J. Mol. Struct.: THEOCHEM, 1995, 330, 347–351 CrossRef CAS.
- M. Casarin, C. Maccato and A. Vittadini, Surf. Sci., 1997, 377–379, 587–591 CrossRef CAS.
- K. Fink, Phys. Chem. Chem. Phys., 2006, 8, 1482–1489 RSC.
- S. A. Farias, E. Longo, R. Gargano and J. B. Martins, J. Mol. Model., 2013, 19, 2069–2078 CrossRef CAS PubMed.
- N. Heshmat, W. L. Christof, M. Martin and W. Yuemin, J. Phys. Chem. C, 2011, 115, 908–914 Search PubMed.
- Y. Zhang, Q. Sun, J. Deng, D. Wu and S. Chen, Appl. Catal., A, 1997, 158, 105–120 CrossRef CAS.
- M. Watanabe, Surf. Sci. Lett., 1992, 279, L236–L242 CAS.
- M. C. Curet-Arana, P. Meza, R. Irizarry and R. Soler, Top. Catal., 2012, 55, 260–266 CrossRef CAS.
- A. Fuchs, E. Kaifer, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2012, 2012, 4020–4028 CrossRef CAS.
- H.-J. Himmel, Eur. J. Inorg. Chem., 2007, 2007, 675–683 CrossRef.
- R. Silaghi-Dumitrescu, M.-M. Uţă, A. Kallay and J. Bodis, J. Mol. Struct.: THEOCHEM, 2010, 942, 15–18 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- C. Xiaobo, S. Shaohua, G. Liejin and S. M. Samuel, Chem. Rev., 2010, 110, 6503–6570 CrossRef PubMed.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- A. Hironori, A. Michele, N. A. John, A. B. Mark, J. B. Eric, T. B. Alexis, E. B. John, C. Carol, D. Eckhard, A. D. David, D. Kazunari, L. D. Daniel, E. Juergen, F. Etsuko, H. G. Dorothy, A. G. William, D. W. Goodman, K. Jay, J. K. Gregory, H. K. Harold, E. L. James, E. M. Leo, J. M. Tobin, M. Keiji, M. N. Kenneth, P. Roy, Q. Lawrence, R.-N. Jens, M. H. S. Wolfgang, D. S. Lanny, S. Ayusman, A. S. Gabor, C. S. Peter, B. R. Stults and T. William, Chem. Rev., 2001, 101, 953–996 CrossRef.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. García, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- W.-N. Wang, F. Wu, Y. Myung, D. M. Niedzwiedzki, H. S. Im, J. Park, P. Banerjee and P. Biswas, ACS Appl. Mater. Interfaces, 2015, 7, 5685–5692 CAS.
- M. Zhong, Y. Ma, P. Oleynikov, K. Domen and J.-J. Delaunay, Energy Environ. Sci., 2014, 7, 1693–1699 CAS.
- T. Wang, R. Lv, P. Zhang, C. Li and J. Gong, Nanoscale, 2015, 7, 77–81 RSC.
- A. Kushwaha and M. Aslam, RSC Adv., 2014, 4, 20955–20963 RSC.
- S. Schenk, J. Notni, U. Kohn, K. Wermann and E. Anders, Dalton Trans., 2006, 4191–4206, 10.1039/b608534b.
- M. Ismael, R. Sahnoun, A. Suzuki, M. Koyama, H. Tsuboi, N. Hatakeyama, A. Endou, H. Takaba, M. Kubo, S. Shimizu, C. A. Del Carpio and A. Miyamoto, Int. J. Greenhouse Gas Control, 2009, 3, 612–616 CrossRef CAS.
- E. M. Mindrup and W. F. Schneider, ChemSusChem, 2010, 3, 931–938 CrossRef CAS PubMed.
- F. D. S. Eirik and F. S. Hallvard, Ind. Eng. Chem. Res., 2006, 45, 2497–2504 CrossRef.
- W. Conway, D. Fernandes, Y. Beyad, R. Burns, G. Lawrance, G. Puxty and M. Maeder, J. Phys. Chem. A, 2013, 117, 806–813 CrossRef CAS PubMed.
- Y. Jiao, A. Du, Z. Zhu, V. Rudolph, G. Q. Lu and S. C. Smith, Catal. Today, 2011, 175, 271–275 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- H. Qin, W. Li, Y. Xia and T. He, ACS Appl. Mater. Interfaces, 2011, 3, 3152–3156 CAS.
- K. Jindal, M. Tomar and V. Gupta, Analyst, 2013, 138, 4353–4362 RSC.
- Z. Xu, S. Chenghua, Y. Hua, C. Zhi Gang, X. Zheng, Y. Delai, L. Gao Qing, L. Xinyong and W. Lianzhou, J. Phys. Chem. C, 2013, 117, 4937–4942 Search PubMed.
- A. P. Bhirud, S. D. Sathaye, R. P. Waichal, L. K. Nikam and B. B. Kale, Green Chem., 2012, 14, 2790–2798 RSC.
- Á. Valdés, Z. W. Qu, G. J. Kroes, J. Rossmeisl and J. K. Nørskov, J. Phys. Chem. C, 2008, 112, 9872–9879 Search PubMed.
- L. Liu, X. Zhao, H. Sun, C. Jia and W. Fan, ACS Appl. Mater. Interfaces, 2013, 5, 6893–6901 CAS.
- Z. Zongyan, L. Zhaosheng and Z. Zhigang, J. Phys. Chem. C, 2013, 117, 6172–6184 Search PubMed.
- J. Wang, W. Luo, J. Feng, L. Zhang, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 16054–16064 RSC.
- H. Ye, G. Chen, H. Niu, Y. Zhu, L. Shao and Z. Qiao, J. Phys. Chem. C, 2013, 117, 15976–15983 CAS.
- Y. Wang, R. Kovacik, B. Meyer, K. Kotsis, D. Stodt, V. Staemmler, H. Qiu, F. Traeger, D. Langenberg, M. Muhler and C. Woll, Angew. Chem., Int. Ed., 2007, 46, 5624–5627 CrossRef CAS PubMed.
- X. Jianping and F. Thomas, J. Phys. Chem. C, 2013, 117, 1804–1808 Search PubMed.
- J. B. Lopes Martins, E. Longo, O. D. Rodríguez Salmon, V. A. A. Espinoza and C. A. Taft, Chem. Phys. Lett., 2004, 400, 481–486 CrossRef.
- Y.-F. Zhao, R. Rousseau, J. Li and D. Mei, J. Phys. Chem. C, 2012, 116, 15952–15961 CAS.
- N. V. Minh and V. N. Tuoc, Proc. Natl. Conf. Theor. Phys., 2010, 35, 101–108 Search PubMed.
- Z.-Y. Zhao, J. Phys. Chem. C, 2014, 118, 4287–4295 CAS.
- Y. Yang, M. N. Weaver and K. M. Merz, J. Phys. Chem. A, 2009, 113, 9843–9851 CrossRef CAS PubMed.
- B. K. Carpenter, J. Phys. Chem. A, 2006, 111, 3719–3726 CrossRef PubMed.
- Y. X. Pan, C. J. Liu, D. Mei and Q. Ge, Langmuir, 2010, 26, 5551–5558 CrossRef CAS PubMed.
- V. Kalamse, N. Wadnerkar, A. Deshmukh and A. Chaudhari, Int. J. Hydrogen Energy, 2012, 37, 3727–3732 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Revision C.01, 2009 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- N. P. Herring, L. S. Panchakarla and M. S. El-Shall, Langmuir, 2014, 30, 2230–2240 CrossRef CAS PubMed.
- Y. Jingyun, L. Changjun, M. Donghai and G. Qingfeng, ACS Catal., 2013, 3, 1296–1306 CrossRef.
- S. Gangarapu, A. T. M. Marcelis and H. Zuilhof, ChemPhysChem, 2013, 14, 3936–3943 CrossRef CAS PubMed.
- J. Ye, C. Liu and Q. Ge, J. Phys. Chem. C, 2012, 116, 7817–7825 CAS.
- X. Sun, X. Cao and P. Hu, Sci. China: Chem., 2015, 1–12, DOI:10.1007/s11426-015-5340-y.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- Y. Yang and D. Cheng, J. Phys. Chem. C, 2013, 118, 250–258 Search PubMed.
- Y.-F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- X. Lü, X. Xu, N. Wang, Q. Zhang, M. Ehara and H. Nakatsuji, J. Phys. Chem. B, 1999, 103, 2689–2695 CrossRef.
- T. Inui and T. Takeguchi, Catal. Today, 1991, 10, 95–106 CrossRef CAS.
- W. H. Doh, P. C. Roy and C. M. Kim, Langmuir, 2010, 26, 16278–16281 CrossRef CAS PubMed.
- G. Peng, S. J. Sibener, G. C. Schatz, S. T. Ceyer and M. Mavrikakis, J. Phys. Chem. C, 2012, 116, 3001–3006 CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10581a |
| This journal is © The Royal Society of Chemistry 2015 |