
Catalytic performance of Co/Zn–Al2O3 Fischer–Tropsch catalysts: a comparative study of zinc introduction methodologies
Abstract
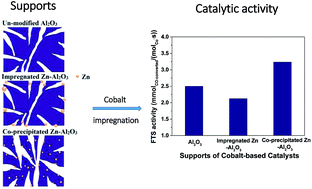
Zinc was introduced into γ-Al2O3 by either co-precipitation or impregnation methods. Zinc introduced by co-precipitation was homogeneously dispersed in the framework of the support, while it was aggregated on the surface of alumina upon addition via impregnation. The co-precipitated prepared Zn–Al2O3 supported cobalt catalyst possessed appropriate pore structure, lower cobalt–support interaction and improved cobalt reducibility, thus showing a significantly enhanced catalytic activity with good stability in Fischer–Tropsch synthesis.
 Please wait while we load your content...
Please wait while we load your content...

