
Regioselective synthesis of novel 2,3,4,4a-tetrahydro-1H-carbazoles and their cholinesterase inhibitory activities†
Abstract
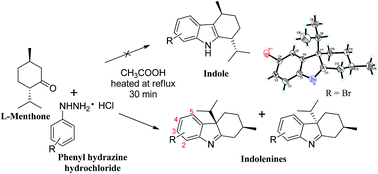
The present study describes the regioselective synthesis of novel 2,3,4,4a-tetrahydro-1H-carbazoles (syn-2(a–q) and anti-3(a–b)) from L-menthone via Fischer indole synthesis. The reaction between L-menthone and different substituted phenyl hydrazines, carried out in acetic acid as catalyst and solvent, yielded a mixture of corresponding 2,3,4,4a-tetrahydro-1H-carbazole diastereomers, instead of the expected indoles. These were characterized by different spectroscopic and single-crystal X-ray diffraction techniques. The isolated single diastereoisomers were further evaluated for inhibitory activities against various enzymes such as cholinesterases, xanthine oxidase, α-chymotrypsin, urease, phosphodiesterase and carbonic anhydrase-II activity, as well as against DPPH radical scavenging and anti-cancer activity against PC-3 cell line. The 2,3,4,4a-tetrahydro-1H-carbazoles show promising inhibitory activities only against acetyl- and butyrylcholinesterase. Moreover, kinetic analyses were carried out for the study of mechanism of inhibition of the active compounds.
 Please wait while we load your content...
Please wait while we load your content...

