
Asymmetric transfer hydrogenation of α-amino β-keto ester hydrochlorides through dynamic kinetic resolution†
Abstract
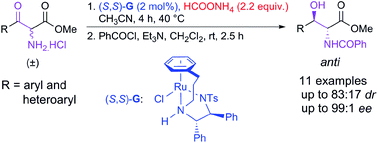
The development of Ru-catalyzed asymmetric transfer hydrogenation of α-amino β-keto ester hydrochlorides is described. The reaction proceeds through dynamic kinetic resolution to afford anti β-hydroxy α-amino esters with good diastereomeric ratios and high enantioselectivities.
 Please wait while we load your content...
Please wait while we load your content...

