
Facile synthesis of AC@TiO2-S with improved visible light photocatalytic activity and recyclability through a controllable sol–gel approach
Zhiying Duan*,
Zhichao Wang,
Chufeng Sun,
Lianbiao Zhao and
Yanbin Wang
Chemical Engineering Institute, Northwest University for Nationalities, Lanzhou 730030, P. R. China. E-mail: zhiyingduan@163.com
First published on 16th June 2015
Abstract
A photocatalyst (AC@TiO2-S) based on active carbon (AC) as a support and TiO2 as active sites has been successfully prepared through a facile, controlled sol–gel approach and characterized by Fourier transform infrared (FT-IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), X-ray powder diffraction (XRD), nitrogen adsorption analysis, transmission electron microscopy (TEM) and scanning electron microscopy (SEM). Importantly, the as prepared AC@TiO2-S exhibits much higher photocatalytic activity than the catalyst AC@TiO2-G (prepared by sol–gel approach) for the degradation of methyl orange (MO) under visible light irradiation (λ > 420 nm). Additionally, this supported catalyst can be easily separated from the reaction system and reused for up to five cycles without significant loss in activity, indicating its excellent recyclability. On the basis of the results, AC@TiO2-S is promising for application in the degradation of other dyes, and the controllable sol–gel method can be applied in other fields.
Introduction
The production of synthetic dyes makes up a large proportion of the chemical industry. Consequently, a lot of non-biodegradable textile dyes are found in textile waste effluents that are discharged into natural streams and water bodies, and these are very harmful and hazardous for the environment. Therefore, a variety of physical, biological and chemical treatment technologies have been explored for the degradation of dyes found in waste effluents. Among them, the photocatalytic degradation of dyes using semiconductors and solar energy for purifying wastewater has been considered a promising method in recent years.1–5Photocatalysis is an ambient-temperature process that can result in the decomposition of organic pollutants and the conversion of solar energy, and presents itself as a green chemistry technology.6 Among the various semiconductors, titanium dioxide (TiO2) has been proven to be one of the most suitable photocatalysts, largely due to its optoelectronic and physiochemical properties. Based on the advantages of high photocatalytic activity, excellent stability and low cost, TiO2 has been widely studied and applied as a photocatalyst in environmental cleaning and energy conversion.7–12 On the other hand, as reported,13 the rapid recombination rate of photogenerated electron–hole pairs within TiO2 may result in its low quantum efficiency. Therefore, if the recombination of charge carriers can be suppressed, the photocatalytic activity of TiO2 will be further enhanced. In the past decades, a large amount of effort has been devoted to modification of the photocatalytic process and improvement of photocatalytic performance.13–18 In particular, carbon–titania hybrid materials have attracted considerable interest because they can potentially offer desirable efficiency for separating electron–hole pairs.19–25 C60 can efficiently generate rapid photoinduced charge separation and relatively slow charge recombination.26 Meng et al.27 prepared a TiO2-type photocatalyst using C60, which had excellent photocatalytic activity for the degradation of Rhodamine B (RhB). Mesoporous TiO2–graphene nanocomposites were synthesized by Liu et al.19 and had good photocatalytic reactivity and tunable photocatalytic selectivity for the decomposition of methyl orange (MO) and methylene blue (MB) in aqueous solution.
Although C60 and graphene as support materials for TiO2 exhibit excellent performances, their high prices make them not ideal for mass production. In our work another carbon material, active carbon (AC), beneficial for enhancing the photocatalytic activity of TiO2 but much cheaper, was used as a support for TiO2. Its large surface area will also be helpful for the photodegradation, because photocatalytic reactions occur on the catalytic surface, which consequently necessitates the prerequisite adsorption of the targeted substrate molecule, and selective photocatalysis on the TiO2 surface is largely dependent on the adsorption selectivity.13
Initially, the TiO2 nanoparticles (NPs) were immobilized by a facile, controllable sol–gel approach. Then the structure of the synthesized catalyst (AC@TiO2-S) was confirmed by corresponding characterization means, and its catalytic activity and reusability were evaluated through the degradation of MO under visible light irradiation (λ > 420 nm), which has become one of the model reactions for testing the photocatalytic activities of various photocatalysts.
Experimental
Synthesis of AC@TiO2-S by a controllable sol–gel method and AC@TiO2-G by sol–gel approach
The synthesis route for AC@TiO2-S and AC@TiO2-G is summarized in Scheme 1. Before preparation of the catalyst AC@TiO2-S, the AC support material (purchased from TianJin Tanggu Binhai chemical plant) needed to be pretreated. The desired amount of AC was dispersed in deionized water and magnetically stirred at room temperature for 10 min, then filtered and dried at 100 °C for 2 h. A solution of ethyl alcohol (2.4 mL) and deionized water (1.0 mL) was adjusted by concentrated nitric acid (65%) until the pH value reached 1, and then this was added dropwise to a mixture of titanium tetrabutoxide (TBT, 5.0 mL), ethyl alcohol (6.0 mL) and acetic acid (1.0 mL), which was kept stirring for 30 min, yielding a light yellow solution. The pretreated AC granules (1.2 g) were added into the above light yellow solution and stirred until the sol was generated and the Tyndall effect was obvious. The product was then filtered and dried at 80 °C for 2 h, and calcined at 400 °C for 4 h in a nitrogen atmosphere. When the gel time was 30 min, the catalyst AC@TiO2-S (AC@TiO2-S2) was eventually obtained. Other catalysts including AC@TiO2-G were also prepared with the same procedure, except that the gel time and calcination temperature were different. The detailed experimental conditions are summarized in Table 1. | ||
| Scheme 1 Schematic diagram of the synthesis route for the AC@TiO2-S and AC@TiO2-G. | ||
| Sample | Amount of TBT used (mL) | Phase | Gel time (min) | Calcination temperature (°C) | BET surface area (m2 g−1) | Ti content (wt%) | C content (wt%) | Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C (atomic ratio) :C (atomic ratio) |
|---|---|---|---|---|---|---|---|---|
| AC@TiO2-1 | 3.5 | Solution | 30 | 400 | 946.6 | 1.1 | 82.7 | 0.33:100 |
| AC@TiO2-S2 | 5 | Sol | 30 | 400 | 940.4 | 3.5 | 77.5 | 1.13:100 |
| AC@TiO2-S3 | 6.5 | Sol | 30 | 400 | 688.3 | 8.7 | 68.8 | 3.16:100 |
| AC@TiO2-S4 | 5 | Sol | 30 | 350 | 940.2 | 3.5 | 77.3 | 1.13:100 |
| AC@TiO2-S5 | 5 | Sol | 30 | 450 | 941.8 | 3.5 | 77.8 | 1.12:100 |
| AC@TiO2-S6 | 5 | Sol | 15 | 400 | 943.2 | 2.5 | 79.6 | 0.79:100 |
| AC@TiO2-S7 | 5 | Sol | 45 | 400 | 922.6 | 4.8 | 75.8 | 1.58:100 |
| AC@TiO2-G | 5 | Gel | 120 | 400 | 523.3 | 11.4 | 63.3 | 4.50:100 |
At the beginning of the experiment, the solution was uniform when the beam was passed through the molecular solution of the precursor of Ti. Invisible light scattering was observed due to mutual interference, and was completely offset. After formation of the colloid, the radius of the dispersed phase particles is generally between 1 and 100 nm, smaller than the wavelength of incident light. A bright red beam could be seen in a perpendicular direction to the incident light – the Tyndall phenomenon. The intensity of scattering light also increased with the increase of particle concentration. Thus during the experiment, colloid concentration could be controlled by the intensity of the scattering light. By controlling the sol, we prepared a series of catalysts (AC@TiO2-S) with well dispersed TiO2 NPs.
Characterization of the catalysts
The structure of the synthesized catalyst AC@TiO2-S was confirmed by characterization. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet NEXUS 670 FT-IR spectrometer with a DTGS detector, and samples were measured with KBr pellets. The powder sample of AC@TiO2-S was analysed through a PHI-5702 multifunctional X-ray photoelectron spectrometer (XPS). X-ray powder diffraction (XRD) measurements were carried out at room temperature and performed on a Rigaku D/max-2400 diffractometer using Cu Kα radiation as the X-ray source in the 2θ range of 20–70°. N2 physical adsorption was carried out on a Micromeritics ASAP 2020 volumetric adsorption analyzer (before the measurements, samples were outgassed at 120 °C for 6 h). The Brunauer–Emmett–Teller (BET) surface areas were evaluated from data in the relative pressure range from 0.05 to 0.20. The total pore volume of each sample was estimated from the amount adsorbed at the highest P/P0 (above 0.99). Pore diameters were determined from the adsorption branch using the Barrett–Joyner–Halenda (BJH) method. The morphology and microstructure of AC@TiO2-S were characterized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The TEM and SEM images were obtained with a Tecnai G2 F30 electron microscope operating at 300 kV and a JSM-5200 JOEL scanning electron microscope, respectively. Elemental analysis was carried out with a conventional combustion method (CHN, varioMLCRO) based on the burn-off mass of the sample and on the analysis of the evolved gases using a thermal conductivity detector. The UV-vis diffuse reflection spectra were obtained for dry-pressed disk samples using a Scan UV-vis spectrophotometer (UV-vis DRS: UV-2550, Shimadzu) equipped with an integrating sphere assembly, using BaSO4 as the reflectance sample. The amount of Ti was determined by ICP-AES (inductively coupled plasma atomic emission, Thermo). The ultraviolet visible (UV-vis) spectroscopy measurements were conducted with a UV2800PC UV-vis spectrophotometer.Evaluation of photocatalytic activity and recyclability
50 mg of granular photocatalyst was suspended in 100 mL of MO aqueous solution (20 mg L−1) and stirred for 2 h before irradiation to ensure that adsorption–desorption equilibrium had been reached. Under visible light irradiation (halogen lamp 250 W, cutoff filter λ > 420 nm), 2 mL aliquots were taken at 30 min intervals during the experiment and centrifuged to remove the powder. The filtrates were analyzed with a UV-vis spectrophotometer.To determine the photocatalytic lifetime, the as-prepared catalyst AC@TiO2-S2 was recovered and reused five times for the decomposition of MO under the same conditions. After each photocatalytic reaction, the aqueous solution was filtered to recycle the AC@TiO2-S2 particles, which were washed and dried at 60 °C for next cycle.
Results and discussion
Characterization of the photocatalysts
The FT-IR spectrum of the catalyst AC@TiO2-S2 is illustrated in Fig. 1. The peak observed at 3425 cm−1 is related to the stretching vibration mode of adsorbed water and hydroxyl groups on the surface of TiO2, and the peak appearing at 1640 cm−1 was attributed to the bending vibration of the O–H bond in the hydroxyl groups and the water absorbed on the solid surface.8,28,29 As reported in the literature, adsorbed water and hydroxyl groups can have a positive effect on the photocatalytic activity.30 The strong intensity of the peaks at 3425 and 1640 cm−1 indicate a high content of adsorbed water and hydroxyl groups in the catalyst, which is beneficial for enhancing the photocatalytic activity of AC@TiO2-S2. The main bands at 400 to 700 cm−1 were attributed to the Ti–O stretching and Ti–O–Ti bridge stretching modes,8,28,29 proving that TiO2 has been successfully modified in the carbon material. Moreover, the peak at approximately 1405 cm−1 reveals the existence of carbonate. | ||
| Fig. 1 FT-IR spectrum of AC@TiO2-S2. | ||
XPS was used to further investigate the chemical elements on the surface. The full-scan XPS spectrum for the catalyst AC@TiO2-S2 is shown in Fig. 2A. Peaks corresponding to C, O and Ti are clearly observed, and the high-resolution XPS spectra of C1s, O1s and Ti2p are depicted in Fig. 2B–D. The C1s peak located at 284.6 eV mainly comes from the support material. The high-resolution XPS spectrum of O1s exhibits an asymmetric peak at 530.3 eV, which was attributed to the lattice oxygen of TiO2,31 and is consistent with previous studies of TiO2. The Ti2p XPS spectrum shows two peaks at 465.0 and 459.3 eV, corresponding to Ti2p1/2 and Ti2p3/2, respectively, which demonstrates the presence of Ti4+ in pure anatase titanium.8
 | ||
| Fig. 2 XPS spectra of AC@TiO2-S2: (A) the survey spectra of AC@TiO2-S2, (B) C1s XPS spectrum, (C) O1s XPS spectrum and (D) Ti2p XPS spectrum. | ||
The XRD patterns of the AC support material and the catalyst AC@TiO2-S are given in Fig. 3. Compared with the pattern of AC, there are five weak diffraction peaks at 25.2°, 38.4°, 47.7°, 55.1° and 63.7° corresponding to the (101), (004), (200), (211) and (204) crystal facets, which were attributed to the single phase, crystalline anatase TiO2.8 No obvious diffraction peak of rutile TiO2 was detected. The diffraction peaks were still observed with the increase of calcination temperature.
 | ||
| Fig. 3 XRD patterns of (a) AC, (b) AC@TiO2-S4, (c) AC@TiO2-S2 and (d) AC@TiO2-S5. | ||
The nitrogen adsorption–desorption isotherms and pore size distributions of the AC support material and the synthesized catalysts are shown in Fig. 4. It can be seen that the AC, AC@TiO2-G and AC@TiO2-S2 exhibit type IV isotherm patterns, demonstrating that they all have a mesoporous structure (Fig. 4A). The BET surface area of AC@TiO2-S2 is 940.4 m2 g−1, which is a little higher than that of AC (931.6 m2 g−1). However, the specific surface area of the catalyst AC@TiO2-G is reduced to 523.3 m2 g−1 (Table 1). What’s more, narrow pore size distributions are observed, centered at 3.75 3.65 and 3.81 nm, respectively for AC, AC@TiO2-G and AC@TiO2-S2 (Fig. 4B). These results indicate that introducing TiO2 to the AC did not destroy the structure of the support material, which can be further confirmed by TEM and SEM analysis.
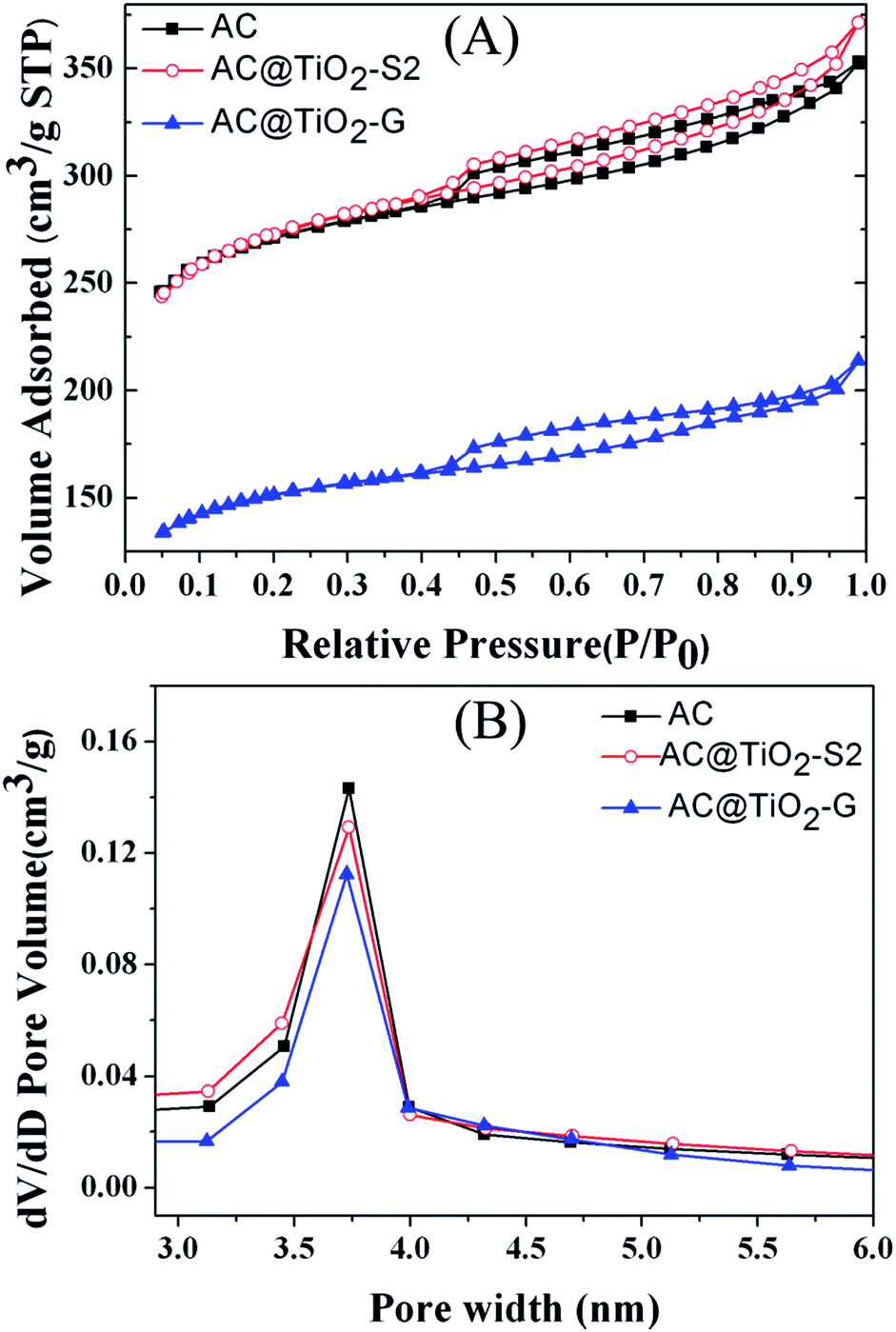 | ||
| Fig. 4 (A) N2 adsorption isotherms and (B) pore size distributions of AC, AC@TiO2-G and AC@TiO2-S2. | ||
The TEM and SEM images given in Fig. 5 provide more information about the morphology and structural features of the synthesized catalyst AC@TiO2-S2. It can be seen directly that the surface of the AC is covered with well-dispersed TiO2 nanoparticles. Obvious aggregation was not observed. The average size of the TiO2 NPs is about 12 nm. In addition, the TiO2 NPs in AC@TiO2-S2 have a perfect crystal structure (Fig. 5B).
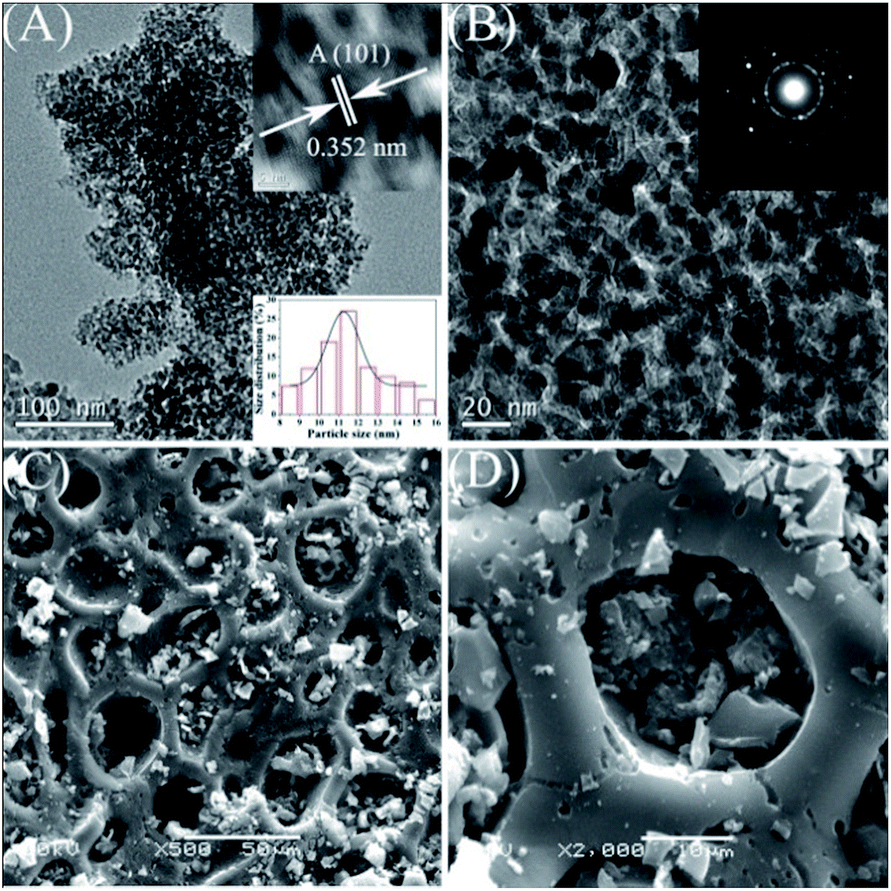 | ||
| Fig. 5 TEM (A and B) and SEM (C and D) images of AC@TiO2-S2. | ||
Evaluation of photocatalytic activity and reusability
With the synthesized catalyst AC@TiO2-S in hand, its photocatalytic activity for the decomposition of MO was evaluated. As is shown in Fig. 6, the process of photocatalytic decomposition of dyes has two steps, the adsorption of dye molecules and the degradation of them catalyzed by AC@TiO2-S2. After adsorption in the dark for 2 h, the sample reached adsorption–desorption equilibrium, and the concentration of MO began to further decrease until the visible light was introduced. The changes in concentration of MO in the reaction system were monitored by measuring the variations in the maximal absorption in the UV-vis spectra at 464 nm (Fig. 6). | ||
| Fig. 6 Successive UV-vis spectra for the degradation of MO in the presence of AC@TiO2-S2. | ||
It is well-known that the preparation conditions have an effect on the performances of catalysts, so the influences of different titanium:carbon ratios, gel times and calcination temperatures in the preparation of the catalysts for photocatalytic activity were investigated. From Fig. 7A, it can be seen that AC has no photocatalytic activity for the degradation of MO. The AC@TiO2-S2 showed the highest photocatalytic activity among the catalysts with different titanium:carbon ratios. In addition, it could be seen that the catalyst prepared at 400 °C (AC@TiO2-S2) exhibited the highest photocatalytic activity, and the conversion of MO was nearly 100% after being irradiated for 150 min (Fig. 7B). A comparison of the photocatalytic activities between the samples prepared at different gel times is given in Fig. 7C. When the gel time was chosen as 30 min, the best conversion of MO was obtained. The relatively close values of degradation rate of MO for AC@TiO2-S2, AC@TiO2-S6 and AC@TiO2-S7 showed that a uniform sol has been generated. When the gel time was about 120 min, the magnetic stirring bar didn’t move and the system turned into a solid gel gradually (AC@TiO2-G). However, the catalytic activity of AC@TiO2-G was greatly reduced compared to AC@TiO2-S2, AC@TiO2-S6 and AC@TiO2-S7. After 180 min, MO had mostly not been degraded.
 | ||
| Fig. 7 Temporal changes in MO concentration in the presence of catalysts prepared (A) at different titanium:carbon ratios, (B) at different calcination temperatures and (C) at different gel times. | ||
Separation and stability are very important for supported photocatalysts. Therefore the reusability of the synthesized catalyst AC@TiO2-S2 was further evaluated and the results are shown in Fig. 8A. It can be seen that the photocatalyst AC@TiO2-S2 was reused in up to five runs without significant loss of activity for the photodegradation of MO, indicating that our catalyst has excellent recyclability. However, the AC had no photocatalytic activity for the degradation of MO and the adsorption of MO for recycled AC reduced. The UV-vis diffuse reflectance spectra (DRS) of the fresh AC@TiO2-S2, used AC@TiO2-S2 (5 times) and AC are thus shown in Fig. 8B. As known, the TiO2 displays a weak absorption intensity ranging from 400 to 800 nm. However, the addition of AC induced increased the light absorption intensity in the UV and visible light regions. Compared with AC, the fresh AC@TiO2-S2 showed significant enhancement of light absorption at a wavelength of 400–800 nm. Furthermore, the AC@TiO2-S2 that had been used 5 times for photodegradation also showed enhancement of light absorption. The results of DRS indicate that the AC@TiO2-S2 has visible light photocatalytic reactivity for organic degradation and has excellent recyclability. TEM was used to get more information about the morphology and structural features of the used AC@TiO2-S2 (5 times), and the TEM images are shown in Fig. 8C and D. Consistent with the fresh AC@TiO2-S2, the TiO2 NPs for used AC@TiO2-S2 were randomly attached on the AC with perfect crystal structures, which further reveals that the AC@TiO2-S2 catalyst has excellent recyclability.
 | ||
| Fig. 8 (A) The lifetime of the catalyst (a) AC@TiO2-S2 and (b) AC for the photodegradation of MO. (B) UV-vis DRS of (a) fresh AC@TiO2-S2, (b) used AC@TiO2-S2 (5 times) and (c) AC. (C) TEM and (D) HRTEM images of used-AC@TiO2-S2 (5 times). | ||
Conclusion
A photocatalyst based on AC as a support and TiO2 as active sites has been successfully prepared through a facile controllable sol–gel approach, and was used in the degradation of MO under visible light irradiation (λ > 420 nm). A high photocatalytic performance was exhibited in the activity evaluation experiments. Moreover, the influence of different titanium:carbon ratios, gel times and calcination temperatures in the preparation of the catalyst for photocatalytic activity were investigated. The synthesized AC@TiO2-S2 had good reusability, evidenced by being extensively recycled for five runs without any substantial loss of activity. On the basis of these results, the photocatalyst AC@TiO2-S2 is promising for application in the degradation of other dyes, and the controllable sol–gel method provides an effective approach for future industrial applications in pollution control and solar energy conversion, owning to its low cost and easy scaling up.
Acknowledgements
This work was supported by the National Science Foundation of China (no. 21463024 and 21462035).Notes and references
- Y. Gao, X.-B. Fan, W.-F. Zhang, Q.-S. Zhao, G.-L. Zhang, F.-B. Zhang and Y. Li, Mater. Lett., 2014, 130, 1–4 CrossRef CAS PubMed.
- Y. Chen, R. Huang, D. Chen, Y. Wang, W. Liu, X. Li and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2273–2279 CAS.
- S. Zhuang, X. Xu, B. Feng, J. Hu, Y. Pang, G. Zhou, L. Tong and Y. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 613–621 CAS.
- G. Zhu, T. Lin, X. Lü, W. Zhao, C. Yang, Z. Wang, H. Yin, Z. Liu, F. Huang and J. Lin, J. Mater. Chem. A, 2013, 1, 9650 CAS.
- W. Zhu, X.-Y. Yang, Y.-H. Li, J.-P. Li, D. Wu, Y. Gao and F.-Y. Yi, Inorg. Chem. Commun., 2014, 49, 159–162 CrossRef CAS PubMed.
- D. H. Wang, L. Jia, X. L. Wu, L. Q. Lu and A. W. Xu, Nanoscale, 2012, 4, 576–584 RSC.
- M. D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231 Search PubMed.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301 RSC.
- Y. Fan, D. Han, B. Cai, W. Ma, M. Javed, S. Gan, T. Wu, M. Siddiq, X. Dong and L. Niu, J. Mater. Chem. A, 2014, 2, 13565 CAS.
- L. Zhang, H. Li, Y. Liu, Z. Tian, B. Yang, Z. Sun and S. Yan, RSC Adv., 2014, 4, 48703–48711 RSC.
- Y. Xie, Y. Li and X. Zhao, J. Mol. Catal. A: Chem., 2007, 277, 119–126 CrossRef CAS PubMed.
- S. Guo, Z. Wu, H. Wang and F. Dong, Catal. Commun., 2009, 10, 1766–1770 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085–4093 CrossRef CAS.
- J.-W. Shi, S.-H. Chen, S.-M. Wang, P. Wu and G.-H. Xu, J. Mol. Catal. A: Chem., 2009, 303, 141–147 CrossRef CAS PubMed.
- X. Hu, G. Li and J. C. Yu, Langmuir, 2010, 26, 3031–3039 CrossRef CAS PubMed.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831 RSC.
- J. Yu, J. Fan and K. Lv, Nanoscale, 2010, 2, 2144–2149 RSC.
- S. Liu, C. Liu, W. Wang, B. Cheng and J. Yu, Nanoscale, 2012, 4, 3193–3200 RSC.
- Y. Sang, Z. Zhao, J. Tian, P. Hao, H. Jiang, H. Liu and J. P. Claverie, Small, 2014, 10, 3775–3782 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Nanoscale, 2011, 3, 3670–3678 RSC.
- Y. Wang, J. Yu, W. Xiao and Q. Li, J. Mater. Chem. A, 2014, 2, 3847 CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- X. Wu, S. Yin, Q. Dong, C. Guo, H. Li, T. Kimura and T. Sato, Appl. Catal., B, 2013, 142–143, 450–457 CrossRef CAS PubMed.
- M. J. Sampaio, C. G. Silva, A. M. T. Silva, L. M. Pastrana-Martínez, C. Han, S. Morales-Torres, J. L. Figueiredo, D. D. Dionysiou and J. L. Faria, Appl. Catal., B, 2015, 170–171, 74–82 CrossRef CAS PubMed.
- M. Melle-Franco, M. Marcaccio, D. Paolucci, F. Paolucci, V. Georgakilas, D. M. Guldi, M. Prato and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 1646–1647 CrossRef CAS PubMed.
- Z. D. Meng, L. Zhu, J. G. Choi, C. Y. Park and W. C. Oh, Ultrason. Sonochem., 2012, 19, 143–150 CrossRef CAS PubMed.
- P. Xu, J. Lu, T. Xu, S. Gao, B. Huang and Y. Dai, J. Phys. Chem. C, 2010, 114, 9510–9517 CAS.
- P. Periyat, D. E. McCormack, S. J. Hinder and S. C. Pillai, J. Phys. Chem. C, 2009, 113, 3246–3253 CAS.
- L. Pan, J. J. Zou, X. Zhang and L. Wang, J. Am. Chem. Soc., 2011, 133, 10000–10002 CrossRef CAS PubMed.
- J. Wang, W. Zhu, Y. Zhang and S. Liu, J. Phys. Chem. C, 2007, 111, 1010–1014 CAS.
| This journal is © The Royal Society of Chemistry 2015 |